
Le Formindep ne soutient pas les récentes prises de positions de François Pesty.

Des traitements plus coûteux, plus vite autorisés, mais de sérieux doutes sur leur efficacité, leur tolérance et les bénéfices qu’ils apportent réellement aux patients
PLAN
Volet 1 – Réforme des ATU : anatomie d’une campagne de lobbying
– Premier acte – Le Sénat sort un rapport d’information bien mal inspiré… 13/06/2018
– Deuxième acte – Le Président reçoit à l’Élysée – 09/07/2018
– Troisième acte – Le 8ème CSIS se tient à Matignon sous la Présidence du Premier Ministre – 10/07/2018
– Épilogue du 1er volet: le LEEM, association des firmes pharmaceutiques, se félicite des grandes avancées françaises – 10/07/2018
– L’alerte de 8 associations de la société civile n’a pas été entendue
– L’évaluation du progrès thérapeutique par la HAS
– L’évaluation du progrès thérapeutique par une revue indépendante
– Des courts-circuits illusoires et dangereux
– Pourquoi faut-il réduire le trop large accès actuel aux immunothérapies ?
– Parlons des cas d’hyperprogression tumorale sous immunothérapie ?
– Extorsion de fonds – Le coût des nouvelles thérapies s’envole
– Pourquoi est-il excessif de parler de guérison à propos de l’HERCEPTIN® ?
– Conclusion du 2ème volet
Volet 3 – Petite revue de presse
Bibliographie
Introduction
La France se targue, et sa Ministre de la santé, Pre Agnès BUZYN la première, de garantir un accès rapide aux innovations thérapeutiques à tous les patients qui le nécessitent. Car notre pays a mis en place un système d’autorisations temporaires d’utilisation (ATU) unique en Europe. Ce système permet de lancer l’utilisation d’un produit, avant même qu’il ne dispose d’une autorisation de mise sur le marché (AMM), d’une évaluation de sa valeur thérapeutique relative par la Haute Autorité de Santé pour le remboursement, ni même d’un prix négocié entre le Comité Économique des Produits de Santé (CEPS) et les industriels((Pour en savoir plus sur le régime des ATU, voir le dossier sur le site de l’ANSM : ici.))
Le 17 octobre, sans véritable débat, l’article 42 du projet de loi de financement de la sécurité sociale (PLFSS) pour 2019 était adopté en commission des affaires sociales, à l’Assemblée nationale. Cet article étend et assouplit encore le régime des ATU.
Cliquer sur l’image ci-dessous pour visionner entre 48:05 et 1:12:58 « l’examen » puis l’adoption de l’article N°42 du PLFSS 2019

Dans un premier volet, nous déroulerons une chronologie politique: la demande de déréglementation des ATU est un cas classique de campagne de lobbying.
Dans un second volet, nous étudierons la réalité de cette « innovation de rupture » alléguée : quelles sont les avancées ? Des patients subissent-ils aujourd’hui réellement une perte de chances faute d’accès à de nouveaux produits ? La stratégie culpabilisante redoutable, utilisée pour pousser à la déréglementation, résiste-t-elle à l’examen des données de la science ? Exemples chiffrés et réaction commune de 8 associations de la société civile permettront de mieux décrypter le mythe de l’innovation de rupture.
Enfin, nous étudierons comment cette campagne de lobbying a investi les médias.
Volet 1 – Réforme des ATU: anatomie d’une campagne de lobbying

Premier acte – Le Sénat publie un rapport d’information bien mal inspiré… 13/06/2018
La MECSS (Mission d’évaluation et de contrôle des comptes de la sécurité sociale), une sous-commission de la commission des affaires sociales du Sénat, a publié le 13 juin un rapport d’information intitulé « Accès précoce à l’innovation en matière de produits de santé »((Ce rapport accessible ici, est téléchargeable en intégralité (pdf 127 pages : ici, ou 1 page html : ici) ou résumé sous forme d’une synthèse (pdf 4 pages : ici). ))
La synthèse du rapport frappe par son lyrisme inhabituel pour un rapport sénatorial : « espoirs formidables pour les patients », « traitements les plus prometteurs », « (dispositif des ATU) unanimement plébiscité par les acteurs de la santé », « les patients bénéficient d’un gain de chances considérable ».
Il est tout aussi frappant de constater que le rapport reprend intégralement tant les critiques du dispositif actuel portées habituellement par les firmes pharmaceutiques, que leurs propositions, qui ne visent qu’à rendre ce dispositif encore plus favorable à leurs intérêts. En voici quelques extraits :
« Le cadre des ATU rencontre aujourd’hui ses limites scientifiques : il ne permet pas les extensions d’indication », [Actuellement les extensions d’indication réclament de la part de l’industriel de déposer un nouveau dossier complet, qui doit être examiné par les différentes instances, EMA, ANSM, HAS, CEPS. D’où de nouveaux délais. Les industriels voudraient obtenir une autorisation conditionnelle qui permettrait de mettre le produit immédiatement sur le marché sans réaliser d’études supplémentaires sur le nouvel organe ciblé…]
« En conséquence, le mécanisme ne garantit pas à tous les patients de recevoir le traitement disponible le plus efficace »,
« La complexité de ce mode de calcul remet en cause la lisibilité du système et pourrait éroder son attractivité » (A propos du « remboursement rétroactif de la différence entre l’indemnité fixée par les laboratoires pendant l’ATU » (prix libre pour la firme) « et le prix du médicament fixé après son AMM, qui repose sur la prévision des volumes de ventes pour les trois années suivant la sortie de l’ATU »
« La procédure en vue de l’admission au remboursement (évaluation HAS et négociation prix avec le CEPS) doit durer en théorie 180 jours,… D’après l’EFPIA (Fédération européenne des entreprises pharmaceutiques) le délai moyen entre AMM européenne et commercialisation est passé à 408 jours (2011-2014) et même à 530 jours (2014-2016), ce qui classe la France loin derrière l’Allemagne et le Royaume-Uni »
« La phase d’évaluation par la HAS est rigoureuse mais ses modalités sont réinterrogées : les AMM, délivrées à un stade de plus en plus précoce sur la base de données immatures, conduisent à une forme de « pari ». Pour les industriels, en résulterait parfois un excès de prudence »
A comparer aux 18 propositions faites par les sénateurs, de façon étonnante toutes favorables aux firmes:

Comment un rapport d’information sénatorial peut-il être amené à reprendre à la lettre l’agenda des firmes pharmaceutiques ? Où sont les biais d’influence ?
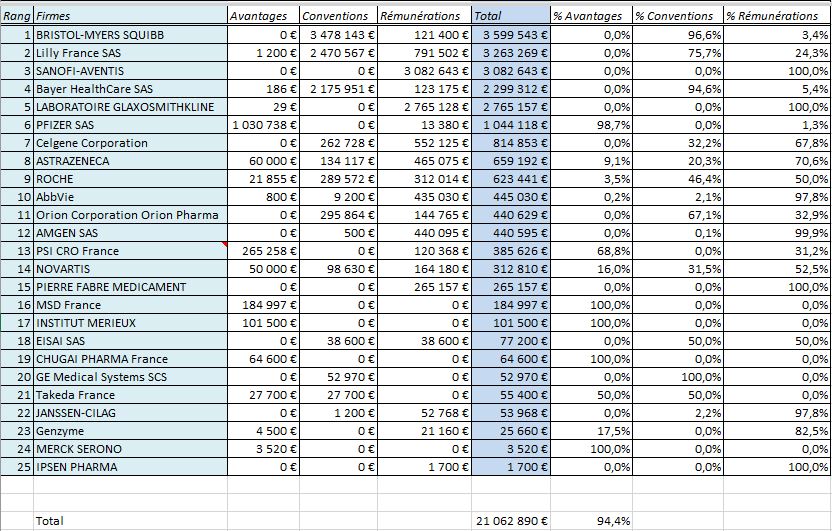
La réponse est en grande partie à trouver du côté des personnes auditionnées par la mission sénatoriale. A de rares exceptions, toutes sont bénéficiaires de ces propositions favorables aux firmes: salariés, responsables et représentants des laboratoires pharmaceutiques; leaders d’opinion, chercheurs, et institutions, rémunérés ou financés par les mêmes firmes. Le manque de diversité et d’indépendance des personnes auditionnées est frappant et explique sans doute l’unanimisme affiché.
Le tableau ci-dessous synthétise, pour chacun des 17 experts auditionnés concernés par la déclaration des liens dans la base Transparence Santé, les nombres et montants totaux d’avantages perçus, les nombres de conventions signées avec les firmes, et les totaux des montants de conventions et de rémunérations. Il faut souligner que la conception même de la base, fixée par arrêté, fait obstacle à ce travail de synthèse((Les rémunérations sont en effet dispersées, sous l’onglet rémunérations, mais aussi en détail d’une convention, ou encore sous l’onglet avantage. Pour les additionner, il aura fallu ouvrir une à une les 552 conventions signées par les firmes avec ces 17 experts entendus par les sénateurs de la MECSS.)) :
Cliquer sur l’image ci-dessous pour télécharger le fichier d’analyse détaillée des liens d’intérêts des 17 experts consultés par les sénateurs (A noter que ce type de fichier peut aussi s’ouvrir avec le logiciel libre « Calc » de la suite OpenOffice)
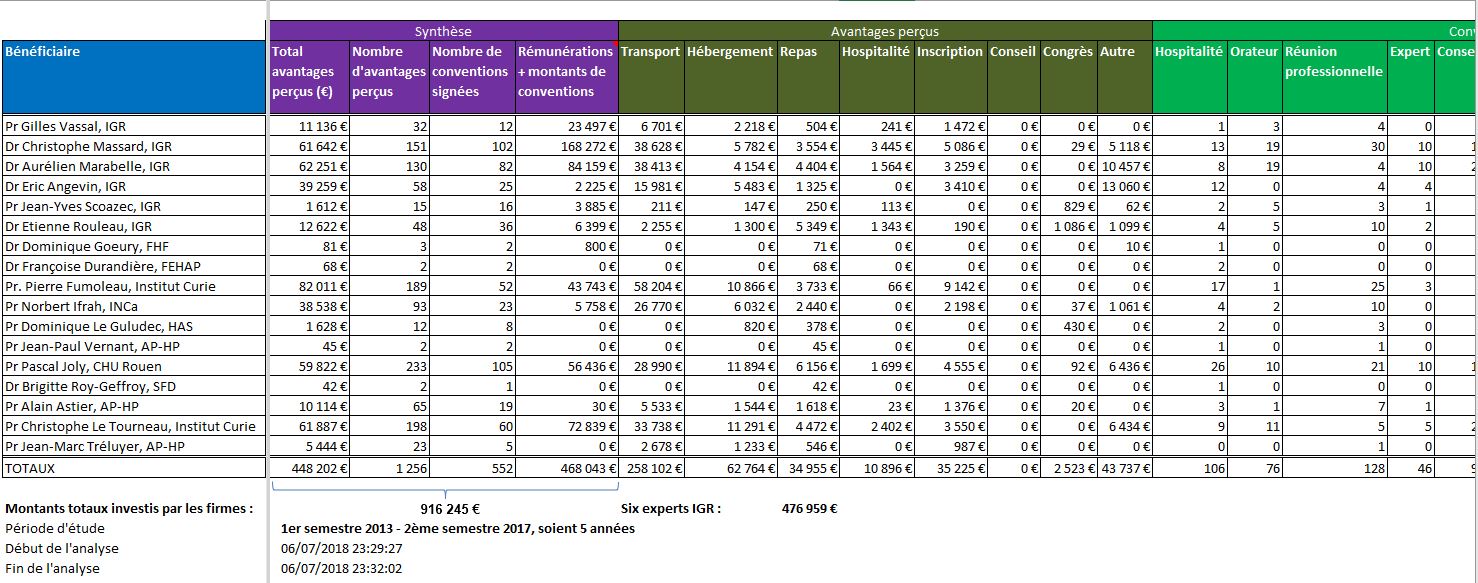
Classement des firmes selon les montants investis sur les 17 experts entendus par les sénateurs
Notons que le Pr. Norbert IFRAH, Président de l’Institut National du Cancer (INCa), n’a plus de liens d’intérêts déclaré dans la base ministérielle depuis sa nomination le 29 juin 2016. Nous savions qu’ils étaient nombreux auparavant, et ceci nous laisse espérer un heureux progrès.
A contrario, l’importance des liens d’intérêts est évidente pour les médecins et responsable des études cliniques de l’Institut Gustave ROUSSY (IGR), ainsi que pour M. le Directeur de l’Institut Curie.
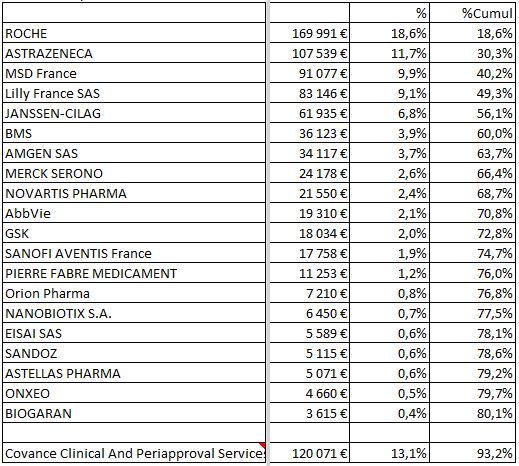
A ces liens personnels de ces experts, il convient d’ajouter les liens qu’entretiennent les institutions dont ils relèvent. Ainsi l’Institut Gustave ROUSSY (IGR) serait bénéficiaire de plus de 22 millions d’euros de rémunérations et dons des firmes:
Cliquer sur l’image ci-dessous pour télécharger le fichier d’analyse des déclarations enregistrées dans Transparence santé
Les 25 firmes ayant déclaré le plus de versements à l’Institut Gustave ROUSSY dans la base Transparence Santé:
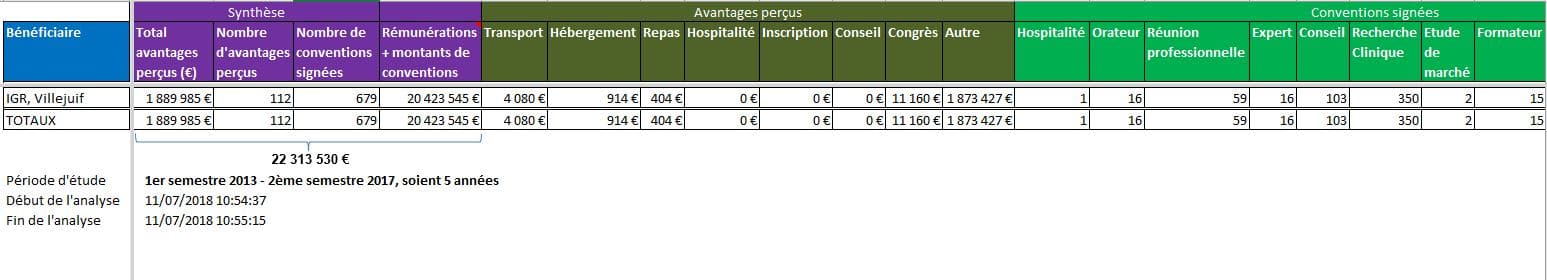
Plus de 7 millions d’euros versés en dons par les firmes à l’Institut Curie, Paris :
Cliquer sur l’image ci-dessous pour télécharger le fichier d’analyse détaillée
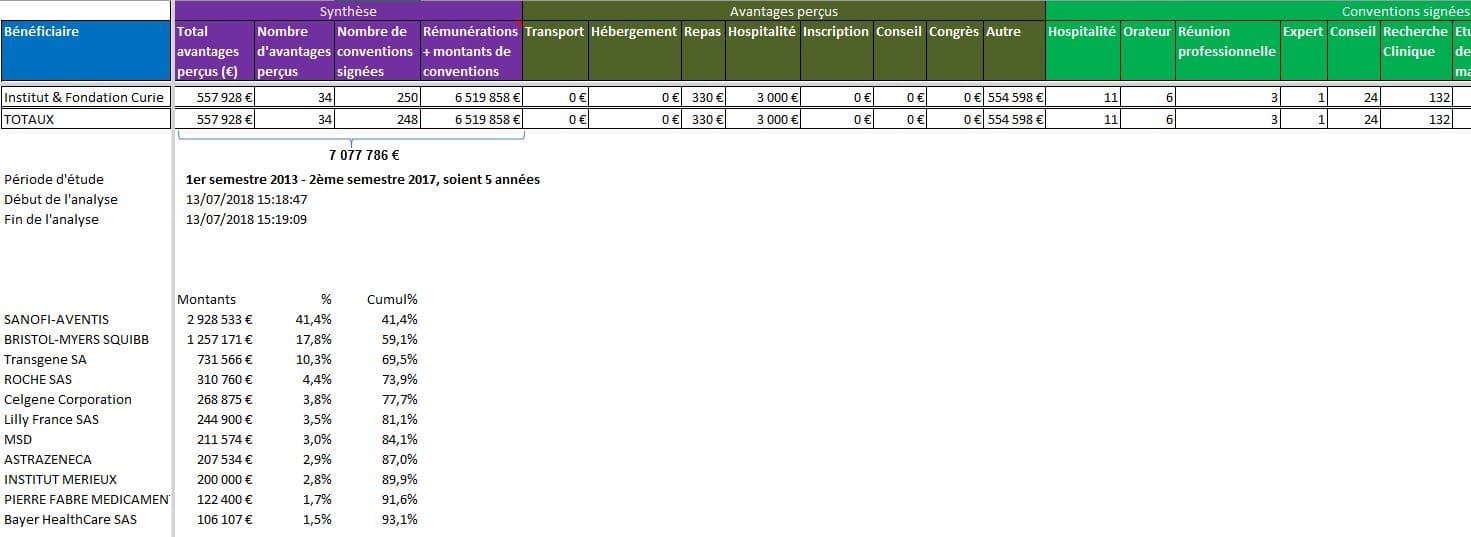
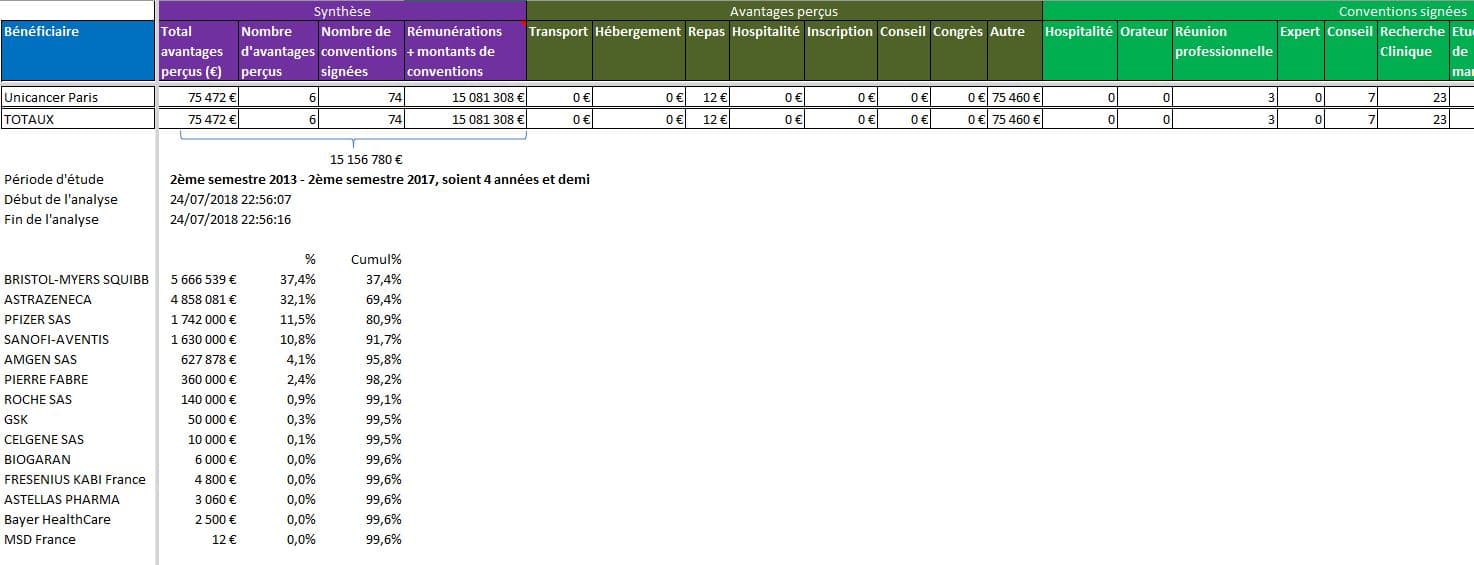
UNICANCER Paris, la Fédération des Centre Régionaux de Lutte Contre le Cancer: plus de 15 millions d’euros de donations:
Cliquer sur l’image ci-dessous pour ouvrir le fichier d’analyse détaillée
Outre ces experts, les sénateurs ont auditionné nombre de dirigeants et lobbyistes des firmes pharmaceutiques concernées((Liste à retrouver sur le site du Sénat : ici.)) :

Ont par ailleurs transmis des contributions écrites :
– Roche
– Bristol-Myers Squibb France
– MSD France
Les « contributions écrites » de ces trois firmes, leaders du marché en matière de nouvelles immunothérapies du cancer, n’ont pas été rendues publiques. Ces firmes produisent et commercialisent respectivement TECENTRIQ®, atézolizumab, OPDIVO®, nivolumab, et KEYTRUDA®, pembrolizumab, trois produits ayant bénéficié du régime d’ATU avant d’obtenir une AMM.
Parmi les firmes auditionnées on remarque également:
– L’association AGIPHARM, mentionnée dans la liste précédente, lobby représentant 14 filiales françaises de laboratoires pharmaceutiques américains (voir : ici),
– Nextep, cabinet de conseil en stratégie spécialisé dans la santé. C’est un lobbyiste du secteur (voir : ici). Son site propose en page d’accueil une interview de Mme Catherine DEROCHE, sénatrice et co-auteur du rapport sénatorial : « Les propositions du rapport sur l’accès précoce aux médicaments innovants – notamment sur les ATU – seront dans le PLFSS » : ici,
– Brigitte CALLES, mentionnée à propos du « Collectif » d’entreprises de biotech (un autre lobby de poids) qui a exercé plusieurs années les fonctions de Secrétaire générale du LIR, « Les laboratoires internationaux de recherche », autre lobby influent regroupant 12 filiales de laboratoires internationaux (voir : ici).
Ainsi, les auditions n’ont accordé de place qu’à des experts en position de conflit d’intérêts, lobbies et industriels. En revanche, les avis indépendants de la Revue Prescrire ou de UFC-Que Choisir, n’ont pas été recherchés, malgré leur manifestation d’intérêt. En témoigne leur contribution, que nous aborderons ultérieurement.
La faible représentation des patients est particulièrement flagrante, pour une réforme de l’ATU qui se présente exclusivement comme une demande de leur part et une défense de leurs seuls intérêts. Notamment les patients atteints de cancers, dont les sénateurs ont beaucoup parlé sans jamais leur donner la parole, et qui semblent avoir servi d’alibi à la déréglementation. Seule représentation de patients auditionnée, France Assos Santé a fait entendre une voix critique, qui tranche avec l’unanimisme allégué par le rapport. Cette commission sert-elle les patients ou se sert-elle d’eux ?
Il est surprenant également que les sénateurs n’aient pas pris en considération les préoccupations, toujours d’actualité, d’une précédente mission du Sénat lui-même face aux dérives des ATU et de l’accès rapide à « l’innovation». Dans les suites du scandale du MÉDIATOR®, tout le circuit du médicament avait été audité. A propos des ATU, le rapport sénatorial de l’époque en 2011 demandait à « modifier le statut des ATU, … afin d’éviter que le titulaire des droits d’un médicament n’utilise cette procédure à seule fin de contourner la procédure d’AMM et le mode de fixation du prix ((Voir plus particulièrement les pages 124-125 : ici)) ».
Dans le même temps, la commission des affaires sociales de l’Assemblée Nationale, dans son projet de Loi ((voir à cet effet la page 22 du rapport relatif au renforcement de la sécurité sanitaire du médicament et des produits de santé : ici)) souligne que « l’ancien système donnait, en effet, lieu à un certain nombre de dérives : les ATU étaient parfois utilisées par les laboratoires pour contourner l’autorisation de mise sur le marché, dans des conditions non encadrées et potentiellement dangereuses pour les patients ».
Le maintien des ATU nominatives et de cohorte s’accompagne d’un « contrôle plus serré », qui s’est traduit dans la Loi relative au renforcement de la sécurité sanitaire du médicament et des produits de santé du 29/12/11.
Retour
Deuxième acte – Le Président reçoit à l’Elysée – 09/07/2018
Dans la torpeur de l’été, quelques jours avant la belle victoire de nos footballeurs, Emmanuel MACRON reçoit le 9 juillet 2018 en grandes pompes à l’Elysée les 25 présidents directeurs généraux (PDG/CEO) des plus grandes firmes pharmaceutiques mondiales ((Sources : Le Point, Le Canard Enchaîné, Le Quotidien du Pharmacien)), dont son ami ((voir leur histoire commune contée par le député François Ruffin, ici)) Serge WEINBERG, PDG de SANOFI.
Ces 25 forment le club fermé du Dolder group, qui tenait une de ses réunions juste avant sa réception par l’Elysée. Des engagements ont-ils été pris ? Peu d’informations ont filtré. Dans une interview, Philippe Lamoureux, DG du LEEM, révèle néanmoins un agenda de dérégulation ambitieux de la part du Président Macron. Il précise que les PDG ont été « impressionnés » par l’engagement du Président, qu’il qualifie de « pro-business« , à les réunir à l’Elysée de nouveau dans un an pour mesurer l’avancement de ce programme.
Le calendrier est idéal et constitue un cas d’école de lobbying : le lendemain débute en effet le Conseil Stratégique des Industries de Santé.
Retour
Troisième acte – Le 8ème CSIS se tient à Matignon sous la Présidence du Premier Ministre – 10/07/2018
Le Conseil Stratégique des Industries de Santé est peu connu du public. Selon le communiqué de presse des services du Premier Ministre (ici) : « Créé en 2004, le CSIS est le lieu où s’élabore une vision stratégique du secteur des industries de santé commune aux pouvoirs publics et aux industriels, et où sont proposées des réponses partagées. Le CSIS associe les principaux dirigeants français et internationaux des industries de santé. La poursuite de cette instance de dialogue entre l’Etat et les industries de santé témoigne du caractère stratégique des industries de santé pour la France et de la volonté des pouvoirs publics de s’inscrire dans un dialogue fructueux, dans un contexte de transformation majeure des industries de santé »
Discours d’Édouard PHILIPPE, Premier Ministre, devant un parterre de dirigeants des firmes à Matignon au 8ème CSIS
Cliquer sur l’image ci-dessous pour visionner l’enregistrement vidéo (24 min 37 s)
« Je ne suis pas scientifique, Madame la Ministre de l’enseignement supérieur, de la recherche et de l’innovation me le fait régulièrement remarquer. Je ne suis pas non plus médecin, Madame la Ministre des Solidarités me le fait régulièrement remarquer, non moi je suis plutôt juriste et littéraire… L’objectif fixé par le Président de la République est d’une grande simplicité à formuler. Nous voulons faire de la France le pays le plus attractif et le plus compétitif d’Europe… Ceux qui sont le plus attentifs au débat politique français, savent que je n’ai pas peur de prôner la décélération sur les routes françaises (rires…). Mais, il m’arrive aussi d’inviter à l’accélération. Pas forcément sur les routes, mais quand c’est nécessaire et notamment quand c’est nécessaire pour mettre à la disposition des français les produits dont ils ont besoin et évidemment les produits dont ils ont besoin lorsqu’ils sont malades… »
«…Vous savez que la France se distingue par un dispositif efficace et reconnu, l’ATU. Grâce à elle, les français sont parmi les premiers en Europe à avoir accès aux nouveaux traitements les plus innovants. 500 ATU ont été délivrées depuis le début de l’année. Il nous semble que ce dispositif marche bien, nous allons donc l’améliorer et l’ouvrir aux extensions d’indications dès le projet de loi de financement de la sécurité sociale de cette année. Concrètement, ça veut dire qu’un médicament déjà présent sur le marché qui soigne, par exemple, le cancer du poumon, pourra se voir accorder une autorisation temporaire pour un autre type de cancer. Un plus grand nombre de malades pourra en bénéficier sans attendre et nous étendrons aux dispositifs médicaux une autorisation analogue à l’ATU ».
Le Premier Ministre n’est ni scientifique, ni médecin, il n’est surtout pas raisonnable. La communauté scientifique alerte sur les risques qu’il y a à accélérer les essais cliniques, réduire « drastiquement » l’évaluation du rapport bénéfice / risque et de l’amélioration du service médical rendu des nouveaux médicaments…
Voici les engagements pris lors du CSIS :
Synthèse des principales mesures annoncées p4
Des délais d’accès au marché accélérés pour favoriser l’innovation
- Réduction des délais d’accès au marché à 180 jours.
- Réduction des délais d’autorisation des essais cliniques respectivement à 60 jours au niveau des CPP, 45 jours pour les médicaments et les DM/DMDIV et 110 jours pour les médicaments de thérapie innovante au niveau de l’ANSM.
- Accélération des dispositifs d’accès précoce à l’innovation pour couvrir plus de patients, pour les médicaments comme pour les dispositifs médicaux.
La mobilisation de la recherche française
- Développement du mandataire unique.
- Facilitation des échanges de personnel pour favoriser les liens public-privé.
- Création du « Health Data Hub », une des plus grandes bases de données de santé au monde.
Une industrie tournée vers l’innovation
- Développement d’une filière de médicaments de thérapie innovante.
- Emergence d’un hub mondial des biotechnologies de demain en France.
- Mobilisation de 2 milliards d’euros de financements publics et privés vers l’innovation (fonds Innobio II et fonds de fonds FABS).
Un dialogue plus stable et plus lisible
- Simplification des règles de régulation du marché des médicaments.
- Visibilité : un plancher minimal de croissance annuelle de 3% pour les médicaments innovants et 0,5% du chiffre d’affaire, correspondant à 1% des dépenses remboursées, pour l’ensemble des médicaments sur trois ans.
- Redéfinition des orientations du Comité Économique des Produits de Santé pour donner toute sa place à la négociation conventionnelle.
- Préparation de la réforme de l’évaluation des médicaments.
Retour
Épilogue du 1er volet: le LEEM, association des firmes pharmaceutiques, se félicite des grandes avancées françaises – 10/07/2018
Le jour même, « les entreprises du médicament » (LEEM) publient un communiqué de presse triomphant: « Nouvelle méthode, nouveau cadre, nouvelle feuille de route… Un CSIS de rupture pour restaurer l’attractivité de la France » (ici), accompagné d’un dossier de presse de 43 pages téléchargeable sur le site du Gouvernement (ici) « Notre ambition pour les industries de santé ».
Le DG du LEEM, Philippe Lamoureux, parle d’un CSIS « amazing« …
Retour
Fin du premier volet

Volet 2 – Fake innovations de rupture
Le mirage des « innovations thérapeutiques » et les dangers du projet gouvernemental
12 juillet 2018 – Déjà auteurs d’un livre blanc livré le 20 juin 2018 « Médicaments et progrès thérapeutique : garantir l’accès, maîtriser les prix – La contribution de la société civile au débat public en France », 8 associations (Aides, France Assos Santé, La Ligue contre le cancer, Médecins du Monde, Prescrire, UAEM, UFC-Que Choisir) émettaient un communiqué de presse commun :
« Conseil stratégique des industries de santé : l’industrie pharmaceutique se réjouit. Contribuables, personnes malades, sommes-nous gagnants ? » qui pose les questions suivantes :
– L’accès accéléré aux nouveaux médicaments sacrifie-t-il la qualité de l’évaluation ?
« Le processus d’évaluation des médicaments est présenté par les industriels comme la principale cause des délais d’accès au marché. Ces retards sont en fait dus au temps de négociation de prix, notamment face aux exigences de prix très élevés des industriels pour des médicaments n’apportant pourtant qu’une amélioration du service médical rendu mineure (IV) ou inexistante (V, soit « absence de progrès thérapeutique »). Les données présentées par les industriels sont souvent trop incomplètes ou ne présentent pas un recul suffisant pour établir qu’ils représentent un progrès tangible pour les patients. Nous sommes particulièrement inquiets sur l’annonce de l’ouverture d’un vaste chantier visant à réformer l’évaluation du médicament qui, au regard des essais fournis par les industriels, mérite justement d’être renforcée ».
– Raccourcir les délais de procédure d’autorisation des essais cliniques, ne constitue-t-il pas un risque pour la protection des personnes ?
– Garantir la croissance du chiffre d’affaire des médicaments remboursables (+3% pour les « inno-vations ») n’est-ce pas courir le risque de compromettre la pérennité de notre système solidaire ?
– Plutôt que de céder face aux prix de plus en plus exorbitants des nouveaux médicaments, ne serait-il pas plus opportun d’exiger une évaluation clinique plus rigoureuse afin de savoir ce qu’ils apportent réellement aux patients ?
Retour
Ne pas confondre innovation et progrès thérapeutique
A entendre les leaders d’opinion et porte-parole de l’industrie, il y aurait extrême urgence à mettre les nouveaux produits sur le marché car ils sauveraient immédiatement des vies. Le PDG de la firme Celgene se vantait récemment: soigner le cancer serait devenu « ironically easy ».
Évaluation officielle du progrès thérapeutique par la HAS : rupture de l’innovation et non pas innovation de rupture
La Haute Autorité de Santé (HAS) n’est pourtant pas de cet avis. Cette « autorité publique indépendante à caractère scientifique » a été créée en 2004. Elle évalue d’un point de vue médical et économique les produits, actes, prestations et technologies de santé, en vue de leur admission au remboursement.
L’évaluation de la valeur thérapeutique des médicaments mis sur le marché se fait au moyen de l’attribution d’un niveau de service médical rendu (SMR, la question posée est de savoir si ce produit répond à un véritable besoin de santé) et d’amélioration du SMR (ASMR, qui permet de situer le produit parmi les alternatives thérapeutiques : apporte-t-il un progrès ?). Voir ici.
La HAS constate peu de progrès parmi les nombreux nouveaux produits et nouvelles indications :
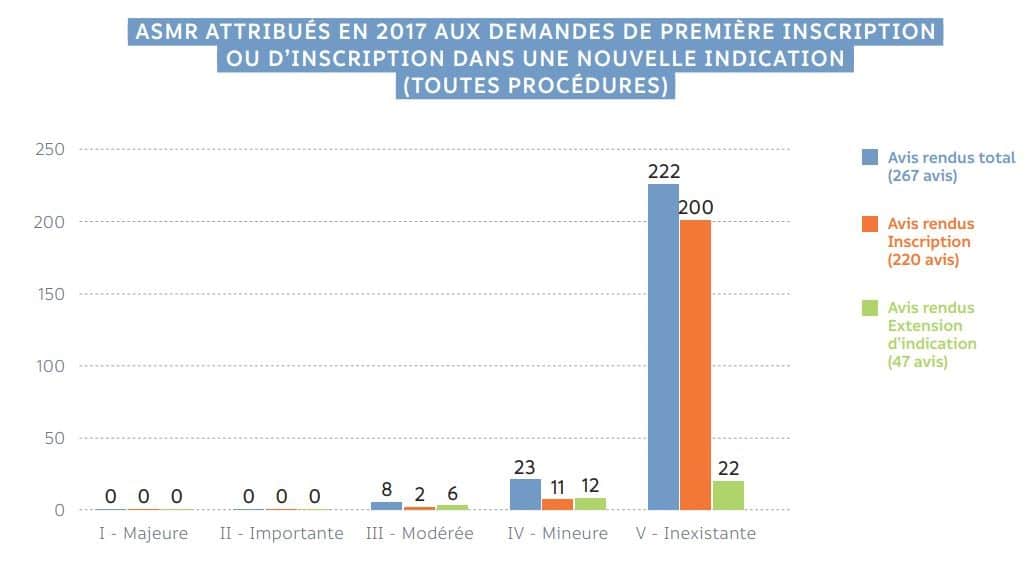
Cliquer sur l’image ci-dessus pour télécharger le rapport d’activité 2017 de la HAS, et voir page 12.
En 2017, sur 53 nouveaux médicaments, seuls 13 apportent un progrès thérapeutique, dont 2 un progrès modéré (ASMR III) et 11, l’essentiel, un progrès mineur (ASMR IV). Aucun nouveau traitement n’a obtenu la mention ASMR I qui est attribuée à un progrès majeur, équivalent à « innovation de rupture ». Aucun nouveau traitement n’a obtenu d’ASMR II correspondant à un progrès important. Trois produits sur quatre n’apportent aucun progrès (ASMR V : progrès nul). 96% apportent au mieux un progrès jugé mineur.
L’année 2017 a-t-elle été une annus horribilis pour le progrès thérapeutique ? Absolument pas : « Depuis 10 ans, on constate une stabilité des appréciations de la commission de transparence… L’ASMR I est attribuée pour des innovations de rupture, qui sont rares. »
Le rapport confirme pourtant l’avalanche de nouveaux produits, particulièrement en oncologie (p28) :
« entre janvier 2015 et décembre 2017, des innovations thérapeutiques sont arrivées à un rythme élevé: parmi les 67 indications évaluées en oncologie ayant obtenu un SMR suffisant, 60 % (40 sur 67) ont obtenu une ASMR I à IV (signant l’apport de ces molécules) et 40 % une absence d’ASMR (signifiant un intérêt similaire aux comparateurs) ».
« …Treize indications ont obtenu une ASMR III grâce à la démonstration d’une supériorité par rapport à un traitement actif sur la survie globale ou sur la survie sans progression ».
80% des nouveaux produits en cancérologie n’apportent donc qu’un progrès au mieux mineur (ASMR IV). L’évaluation de la Haute Autorité de Santé tempère les discours lyriques sur le « retour de l’innovation » (dont on ne nous avait pourtant jamais dit qu’elle était partie).
Peut-être l’agence française est-elle particulièrement sévère ? C’est peu vraisemblable. Les agences d’évaluation des technologies de la santé (HTA : Health Technology Assessment) homologues de la Haute Autorité de Santé partagent en effet ce constat. Ainsi, sur la même période, l’agence canadienne CEPMB a-t-elle jugé que 82% des produits présentaient un progrès « minime ou nul ». Quant à l’agence allemande IQWIG, elle estime que 73% des produits ne démontrent aucun progrès, voire une régression (l’IQWIG distingue en effet 1% de produits qui représentent une régression nettement démontrée par rapport aux thérapeutiques existantes).
Retour
Évaluation du progrès thérapeutique selon une revue indépendante de l’industrie
Les évaluations indépendantes de la revue Prescrire, que les leaders d’opinion porte-parole de l’industrie aiment à présenter comme extrémiste, apparaissent dès lors concorder avec les évaluations officielles. Selon la revue, seuls deux produits qu’elle a évalués en 10 ans ont représenté un progrès majeur, tandis que 15% des produits n’auraient pas mérité d’être mis sur le marché.
Le fait que le terme de « progrès thérapeutique » ait disparu au profit de celui d’ « innovation », selon les éléments de langage de l’industrie pharmaceutique, est en lui-même révélateur. Pierre Chirac, de la Revue Prescrire, l’a fort bien exprimé lors de son audition par l’Assemblée Nationale :
Cliquer sur l’image ci-dessous pour lancer la vidéo (7 min 40 s)

« En fait,… innovation veut juste dire nouveau. Ça ne veut pas dire progrès, ce dont probablement peu de soignants et surtout peu de patients se doutent. Mais, pour les gens dans les agences, c’est évident, innovation ça veut dire nouveau. La plupart des innovations n’apportent pas de progrès tangible pour les patients et certaines s’avèrent de véritables régressions thérapeutiques. Ce constat regrettable, Prescrire n’est pas seul à le faire, et, par exemple, des cancérologues des États-Unis ont publié une étude montrant qu’avec les 71 médicaments anticancéreux pour des tumeurs solides mis sur le marché entre 2002 et 2014,…, l’allongement de la durée de vie n’avait été que de 2 mois seulement en moyenne… Est-ce que les patients sont bien conscient de cela ? Probablement pas. Est-ce que les responsables politiques sont conscient de cela ? Probablement pas non plus ».
AMM : des exigences d’efficacité de plus en plus réduites
Comment en est-on arrivé à mettre de façon si indiscriminée les produits sur le marché ? Les exigences de l’Autorisation de Mise sur le Marché (AMM) sont aujourd’hui si basses qu’il semble difficile de les abaisser davantage.
L’Agence Européenne du Médicament n’a jamais eu pour mission de s’assurer que les produits qu’elle mettait sur le marché constituaient une avancée pour les patients ou les systèmes de santé. Son rôle se limite à contrôler que les risques liés au produit restent acceptables comparés à ses bénéfices. Un rôle que l’agence doit remplir de surcroît « par des moyens qui ne puissent pas freiner le développement de l’industrie pharmaceutique » (Directive 2001/83/CE).
Il n’existe pas de critère de ce qui est jugé un rapport bénéfice/risque « acceptable », et le seuil d’acceptabilité n’a jamais fait l’objet d’un débat public, et est laissé à quelques experts de l’EMA. Au terme d’un long processus de capture du régulateur, l’Agence Européenne du Médicament (EMA) est aujourd’hui réduite à l’état de chambre d’enregistrement. Pourtant, évaluer l’effet des produits n’est-il pas précisément le rôle de l’Agence ?
Dans une synthèse consultable librement, Prescrire estime qu’ « au fil des années, il se confirme que l’évaluation des médicaments en vue d’obtenir une autorisation de mise sur le marché (AMM) est trop souvent bâclée ».
La Haute Autorité de Santé, dans son premier rapport de prospective, évoque des « données très préliminaires ou partielles », un « manque de robustesse ». Les nouvelles technologies sont en effet mises sur le marché de plus en plus tôt après leur développement, parfois sans que des données suffisantes ne soient disponibles pour lever l’incertitude sur leurs effets »
Une analyse plus précise de la méthode d’évaluation par l’EMA des traitements du cancer ne laisse pas d’inquiéter.
Dans le bilan de l’année 2017, Prescrire consacre un paragraphe à l’oncologie :
« Médicaments en cancérologie : illustration des dérives
28 nouveautés médicamenteuses sur 92 analysées en 2017 ont concerné des médicaments utilisés dans les cancers. Certaines de ces AMM ont été octroyées sans disposer d’essais comparatifs….
Parmi ces 28 AMM de médicaments en cancérologie, 20 ont été accordées sur la base d’un seul essai clinique, souvent de qualité méthodologique médiocre car non comparatif ou avec des biais liés à son caractère non aveugle ; ou sur la base de critères biologiques ou radiologiques, pas forcément corrélés à un allongement de la durée de vie ou à une amélioration de la qualité de vie.
En somme, dans le domaine de la cancérologie, on note beaucoup de commercialisations sur un marché rendu très attractif par sa facilité d’accès pour les firmes, à des prix exorbitants et déconnectés du progrès thérapeutique ou des coûts de recherche et développement ».
Se passer des données de la science
L’AMM ne jouant plus son rôle de filtre, le tri des produits se reporte de façon croissante sur les agences HTA, qui sont pour cette raison la bête noire des industriels, désormais suivis en cela par les gouvernements. Mécontente des avis peu enthousiastes des agences HTA, qui persistent à jouer leur rôle de garde-barrière vis-à-vis de la prise en charge de ces produits par la solidarité nationale, l’industrie pharmaceutique a commencé par dénigrer ces agences. L’instrumentalisation indécente du cas d’une jeune femme présentée comme privée d’un traitement « miracle » par une décision « bureaucratique » relève de cette stratégie.
Cédant à cet odieux chantage affectif, les politiques ont multiplié les voies de contournement (ex: le fonds cancer britannique, la liste en sus et les ATU en France) pour permettre la prise en charge par les régimes sociaux de produits pourtant recalés par les agences HTA, ou pas encore examinés faute de données scientifiques.
Il est temps d’aborder maintenant les raisons pour lesquelles il est dangereux de vouloir accélérer l’évaluation et l’accès aux nouveaux médicaments, comme le souhaitent les industriels et le gouvernement. Mais aussi pourquoi les nouvelles immunothérapies et thérapies « ciblées » du cancer n’offrent pas autant de valeur aux patients que voudraient bien nous faire croire toutes celles et tous ceux qui ont d’autres intérêts que celui de la santé des personnes.
Retour
Des courts-circuits illusoires et dangereux
Les USA ont une avance qu’il ne faudrait pas trop leur envier, en matière de procédures accélérées d’autorisation de mise sur le marché pour les « innovations » ; Les chercheurs sérieux et indépendants ont alerté sur les risques de tels dispositifs dans les plus grandes revues médicales. Voir la bibliographie proposée en fin d’article. Le pire étant à venir avec la Loi promulguée par Donald TRUMP le 1er juin 2018, « The Right to Try Act », que l’on peut traduire par « le droit d’essayer », qui donne la possibilité aux patients américains en phase terminale de prendre des médicaments non encore approuvés par la FDA.
Les autorisations expéditives de médicaments, posent le problème d’une information manquante sur leur sécurité et leur efficacité, qui potentiellement majore les risques pour les patients 1-29. Ces médicaments sont évalués le plus souvent sur la base de résultats préliminaires portant sur des critères de jugement indirects, comme le taux de réponse de la tumeur ou la survie sans progression de la tumeur, ou sans métastase. Ce qui n’a évidemment pas la même valeur informationnelle pour décider d’un traitement que les critères les plus pertinents que sont la survie globale ou la qualité de vie. La qualité de vie est d’ailleurs très rarement étudiée dans les essais thérapeutiques. Les anticancéreux présentent le plus souvent des effets indésirables graves. Or, les études de phase 3 sont conçues pour évaluer l’efficacité, et non la tolérance. Ainsi, les effets indésirables les plus sévères, moins fréquents, n’apparaissent qu’après utilisation de ces médicaments sur des cohortes plus importantes de patients. Les médicaments autorisés sur procédure accélérée par la FDA font davantage l’objet ultérieurement d’alertes de sécurité 6. Des retards inacceptables sont constatés dans le retrait du marché de médicaments autorisés après qu’ils aient été associés à des décès 11.
La littérature rapporte de façon récurrente qu’un grand nombre d’anticancéreux n’ont jamais démontré qu’ils allongeaient la vie. Des études conduites sur les médicaments autorisés (FDA) dans le cancer aux USA 12, 13, 15, 16, et en Europe (EMA) 14, 17-20, montrent que plusieurs années après leur autorisation la plupart des médicaments anticancéreux n’ont toujours pas démontré un bénéfice sur la survie globale ou l’amélioration de la qualité de vie. De plus en plus souvent des AMM sont délivrées conditionnellement sur la base de critères de jugement indirects 12, 13, 16, 21, 23 sous réserve que l’industriel s’engage à réaliser une étude post-AMM pour prouver un bénéfice sur la survie globale. Les résultats de ces études sont très souvent négatifs, voire jamais publiés ou les études ne sont jamais réalisées. Ces médicaments ne sont malheureusement pas retirés du marché. L’autorisation accélérée de mise sur le marché devrait être contrebalancée par un retrait accéléré lorsque le médicament ne confirme pas un bénéfice sur la survie globale ou la qualité de vie dans les essais post-AMM 22. Les agences sanitaires sont très laxistes sur le sujet. La Cour des Comptes américaine a déjà soulevé ce problème 23,24.
Selon des chercheurs, les conséquences inattendues du prix exorbitant des nouveaux anticancéreux sont la poursuite de gains marginaux (« progrès incrémental »), mais également l’adoption d’une mentalité « mee-too », que l’on peut traduire par « moi aussi je veux ma part du marché », et qui bloque toute innovation réelle et toute créativité 25. La tendance est de voir très (trop) rapidement appliqués à la pratique courante ces médicaments approuvés selon une procédure accélérée en dépit des risques inhérents à leur mauvaise connaissance des risques qu’ils font encourir aux patients 26.
L’utilisation par les leaders d’opinion, hélas reprises dans les médias, de superlatifs inappropriés au sujet de ces nouveaux médicaments est un lieu commun 27. Les avantages et rémunérations perçus par les oncologues de la part des firmes qui commercialisent des anticancéreux encore protégés par des brevets et donc plus coûteux, coïncident avec leur prescription préférentielle 28. De surcroît, les prix astronomiques observés ces dernières années n’ont aucune justification 29. Aux USA une équipe de chercheurs vient tout juste de publier les résultats de leur étude systématique dans la base de données en open data (ici) sur les liens d’intérêts financiers des 344 oncologues américains mentionnés comme auteurs et co-auteurs dans 43 essais cliniques publiés concernant tous les médicaments d’oncologie et d’hématologie approuvés par la FDA entre le 1er janvier 2016 et le 31 août 2017. 263 oncologues-auteurs (76,5%) ont reçu au moins 1 paiement, pour un montant total cumulé de 217 millions de dollars. 110 oncologues (32%) n’ont pas déclaré tous leurs liens d’intérêts financiers. Un quart des oncologues-auteurs n’ont reçu aucun paiement, ce qui montre qu’il est possible de participer à des essais cliniques tout en les refusant.
Retour
Pourquoi faut-il contrôler l’accès aux
« nouvelles immunothérapies » et « thérapies ciblées » du cancer ?
Ces médicaments* peuvent s’avérer efficaces avec de substantiels gains de survie, mais uniquement chez un très petit nombre de patients, alors qu’ils sont prescrits à tous. Il en résulte que si l’on peut parfois s’émerveiller devant une différence de médianes de survie entre le groupe de patients traités par ces médicaments et un comparateur actif, il aura fallu pour cela traiter inutilement 83% à 94% des patients qui eux n’ont bénéficié d’aucun allongement de leur vie. A contrario, 14% des patients auront malheureusement subi une « hyperprogression tumorale » associée à un raccourcissement significatif de leur vie !
(*) : En l’espèce, un travail de recherche présenté au colloque 2018 de Bobigny a concerné les six molécules suivantes : KEYTRUDA®, pembrolizumab, MSD ; OPDIVO®, nivolumab, BRISTOL-MYERS SQUIBB ; TECENTRIQ®, atézolizumab, ROCHE ; YERVOY®, ipilimumab, BRISTOL-MYERS SQUIBB ; TAGRISSO®, osimertinib, ASTRAZENECA ; CABOMETYX®, cabozantinib, IPSEN PHARMA ; ainsi que la combinaison de deux d’entre-elles (nivolumab + ipilimumab), dans onze essais cliniques « pivots », portant sur 4 localisations différentes de cancers avancés, (cancer bronchique non à petite cellule, mélanome malin, carcinome rénal, cancer épidermoïde de la tête et du cou) en 1ère ou en 2ème ligne. La présentation diapositive sonorisée de ce travail est téléchargeable : ici. Et le poster scientifique : ici.
Pour comprendre comment l’on parvient à ces résultats beaucoup moins flatteurs, il est nécessaire d’aborder deux modes de représentation des résultats des essais clinique en cancérologie préférables à celui de la différence des médianes de survie, laquelle peut être très trompeuse si elle est le fait d’un tout petit nombre de patients.
Il s’agit en premier lieu du « gain absolu de survie ». Il est égal à la différence des taux de survie observés dans les deux groupes de l’essai clinique aux différentes durées de suivi.
A partir de ce gain absolu de survie il est aisé de calculer le « nombre de patients à traiter » (NNT 30-32 pour « number needed to treat » en anglais) pour éviter un événement (ici un décès quelqu’en soit la cause, lorsque l’on s’intéresse à la survie globale). Le NNT est égal à l’inverse du taux absolu de survie.
Pour le KEYTRUDA®, pembrolizumab, l’un des produits les plus convaincants, autorisé en France en 1ère ligne dans certains cancers bronchiques à non petites cellules (CBNPC), dès lors que plus de 50% des cellules tumorales expriment le gène muté (PD1/PD-L1), les gains absolus de survie observés à 6, 12 et 24 mois de suivi (ici), étaient respectivement de 9,8% ; 15,5% et 17,0%. Le nombre de patients à traiter pour éviter un décès à 6 mois dans le groupe pembrolizumab par rapport au comparateur actif est donc de 1/0,098 = 10. Il faut donc traiter 10 patients pour éviter un seul décès à 6 mois. 9 patients sur 10 (90%) auront donc été traités pendant cette période sans gain de survie. De la même manière, les NNT pour éviter un décès à 12 et 24 mois sont de 6. Il aura donc fallu traiter 6 patients pendant 12 ou 24 mois pour éviter un seul décès. 5 patients sur 6 (83%) auront donc été traités 12 ou 24 mois sans gain de survie.
Formulé différemment, les patients dont plus de 50% des cellules tumorales expriment le gène PD1/PD-L1, n’ont pas plus de chance d’obtenir un sursis de 6, 12, 18 mois, on ne sait pas très bien encore, que de faire un six en lançant un dé…
Hélas, peu d’oncologues tiendront ce langage de vérité, à laquelle devrait avoir droit tout patient, préférant affirmer qu’ils lui offrent « le meilleur traitement » ou « le traitement le plus efficace » disponible.
Alors que ces médicaments sont présentés comme des « innovations de rupture », inaugurant la médecine dite « de précision », les proportions de cellules tumorales exprimant la mutation génomique, en l’occurrence, PD1/PD-L1 pour les 3 premières molécules citées, ne sont pas utilisées alors qu’elles devraient l’être, pour définir des seuils pertinents d’éligibilité au traitement. Les firmes n’ont fait aucun effort pour sélectionner les seuls patients répondeurs. Elles n’y avaient d’ailleurs aucun intérêt, puisque leur objectif est de traiter le plus de patients possibles… Les autorités sanitaires, FDA, EMA, ANSM, HAS, INCa ont été très complaisantes. Dans le seul cas où un seuil d’éligibilité au traitement a été défini, le KEYTRUDA® en 1ère ligne dans le CBNPC, celui-ci, nous venons de le voir, est notoirement insuffisant.
Retour
S’agissant des nouvelles immunothérapies anti-PD1/PD-L1, les résultats d’une étude récente publiée dans le JAMA Oncology 34, sont encore plus préoccupants, car près de 14% des patients auront une survie réduite significativement, suite à une « hyperprogression de leur tumeur » :
Dans une cohorte de 406 patients traités en deuxième ligne par l’une des 4 immunothérapies suivantes : nivolumab, pembrolizumab, atezolizumab, ou durvalumab (DURVALUMAB®, Astrazeneca, sous ATU, prix libre et gardé secret), une hyperprogression de la tumeur (HPD) a été observée chez 13,8% des patients et associée à une diminution significative de la survie globale. La définition de cette accélération de la croissance tumorale était jugée « conservatrice » par l’un des auteurs interviewé par la revue médicale 35, qui n’excluait pas une fréquence plus importante avec une définition plus large (Ce chiffre de 13,8% a été obtenu en prenant en compte un accroissement de la vitesse de croissance tumorale de +50% après la début du traitement par rapport à deux scanners réalisés avant). Cet auteur estime aussi que cette accélération de la croissance tumorale et la surmortalité associée doivent s’observer avec une fréquence probablement similaire chez les patients traités en 1ère ligne…
Retour
Extorsion de fonds :
Ces produits sont proposés à des prix exorbitants : KEYTRUDA®, pembrolizumab, représente un coût de traitement mensuel de 7.753,23 € et annuel de 93.000 € en 1ère ligne dans le CBNPC. Le coût supplémentaire par rapport au comparateur actif (sels de platine + pémétrexed, la plus coûteuse des 3 chimiothérapies au choix de l’investigateur dans l’étude KEYNOTE 024) pour éviter un décès au bout de 24 mois de traitement est de 680.000 € (Il faut traiter 6 patients pendant 2 ans).
Ce même médicament, administré en 2ème ligne présente un surcoût de traitement par rapport au comparateur actif (docétaxel dans l’étude KEYNOTE 010) pour éviter un décès au bout de 12 mois est de 831.000 € (Il est nécessaire de traiter 12 patients pendant un an)
Le pembrolizumab, objet du débat sur France Médicale que nous analyserons dans le 3ème et dernier volet de l’article, présente un coût de traitement de 47.000 euros par patient et par an dans l’indication du cancer métastasé de la vessie, alors que 87,5% des malades traités dans l’essai clinique qui lui a permis d’obtenir une autorisation de mise sur le marché dans cette nouvelle indication, n’obtiennent aucun gain de survie après un an de traitement par rapport au comparateur actif. Une surmortalité est même observée dans les deux premiers mois de traitement (probablement liée à l’hyperprogression tumorale dont nous venons de parler), comme le souligne l’avis de la commission de la transparence HAS (ici).
Retour
Pourquoi est-il excessif de parler de guérison à propos de l’HERCEPTIN® dans les cancers du sein ?
Le trastuzumab, HERCEPTIN®, laboratoire ROCHE, souvent considéré comme exemplaire en matière de « thérapies ciblées » du cancer, a donné des résultats mitigés dans les essais cliniques. Au point que parler à son propos de « guérison » comme n’a pas hésité à le faire un oncologue leader d’opinion (à retrouver dans le 3ème volet de l’article) parait totalement excessif. Et voici pourquoi :
Environ 20 à 25% des femmes atteintes d’un cancer du sein présentent une tumeur qui surexprime le gène HER2 et sont donc éligibles depuis le début des années 2000 au traitement par trastuzumab.
On distingue principalement deux situations :
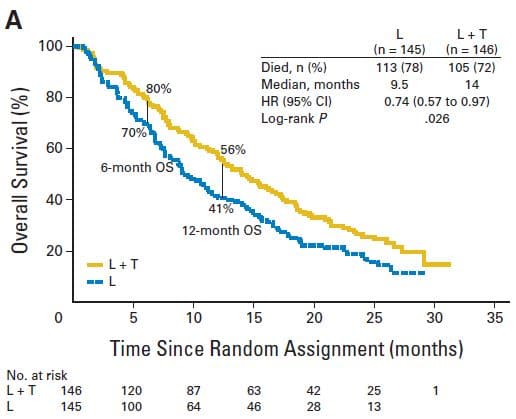
La première est représentée par le cancer du sein métastasé, de mauvais pronostic, pour lequel la durée de suivi dans les essais pivots publiés 37-39 ne dépasse pas 24 ou 30 mois pour le trastuzumab associé à un taxane (paclitaxel ou docétaxel), Par exemple, pour le premier essai clinique publié en 2001 37, qui a inclus le plus grand nombre de patientes (469), au bout de 30 mois, il ne restait que 36,6% de survivantes dans le groupe traité par chimiothérapie + trastuzumab contre 29,5% de survivantes avec la seule chimiothérapie. Soit un gain absolu de survie globale de 7,1%. Ce qui signifie qu’il faut traiter 14 patientes pendant 30 mois avec du trastuzumab pour avoir une survivante de plus par rapport au groupe qui ne reçoit que la chimiothérapie. Nous aurons donc 13/14, soit 93% de patientes traitées inutilement pendant 30 mois, sans aucun gain de survie globale. La seconde étude publiée en 2005 38, n’a abouti qu’à un gain absolu de survie de 4% après 24 mois, soit un surtraitement de 96% des patientes incluses dans l’essai. Une troisième étude 39 ne sort pas statistiquement significative sur la survie globale. Deux autres essais ont été réalisés en associant au trastuzumab un inhibiteur de l’aromatase 40-41 (anastrozole ou létrozole). L’un donne aussi un gain absolu de survie de 7%, l’autre aucun gain significatif. Dans une cinquième étude le trastuzumab était associé au lapatinib, une autre thérapie ciblée et comparé au lapatinib seul chez 291 femmes 42. Le gain absolu de survie est de 10% à six mois et de 15% à un an (graphique ci-dessous). Heureusement que les résultats n’ont pas été publiés à 24 mois car ils étaient beaucoup moins favorables. Malheureusement, les pentes des courbes de survie (dites de « KAPLAN-MEIER ») ne laissent guère de doute sur l’issue de cette grave maladie, que les patientes aient été ou non traitées par trastuzumab.
Une revue systématique de la collaboration Cochrane 43 publiée en 2014, ne retenait que 7 études dans le cancer du sein métastasé traité par trastuzumab seul ou associé à une chimiothérapie et / ou un traitement hormonal et / ou une thérapie ciblée versus le même traitement sans trastuzumab, aucune réalisée en aveugle. Seules 5 de ces études avaient publié des données de survie globale. Prises individuellement aucune ne montrait de différence significative. En combinant les données des 5 études, la Collaboration Cochrane observe un allongement de la survie globale de 5 à 8 mois et un Hazard ratio de 0,82 avec un intervalle de confiance à 95% compris entre 0,71 et 0,94. Soit un gain absolu de survie de 18%. A contrario, le risque d’insuffisance cardiaque congestive est multiplié par 3,5 avec le trastuzumab.
A noter dans une étude de cohorte portant sur 353 patientes traitées en 1ère ligne par trastuzumab pour un cancer avancé 44, que les femmes qui avaient reçu du trastuzumab au préalable en chimiothérapie adjuvante ou néo-adjuvante (traitement avant la chirurgie, visant à réduire la taille de la tumeur pour l’extirper plus facilement) lors de la prise en charge précoce de leur cancer, présentaient après apparition de métastases une survie globale réduite de 8 mois par rapport aux femmes naïves de trastuzumab. Une étude que le laboratoire ROCHE s’est bien gardé de verser au dossier lors de la dernière évaluation par la commission de la transparence de la HAS (ici)…
Une seconde situation concerne le cancer du sein précoce :
Il s’agit d’une pathologie d’évolution plus lente. Le traitement par trastuzumab est alors associé à une chimiothérapie dite « adjuvante », car proposée après la chirurgie, avec ou sans radiothérapie. Bien que les essais cliniques pivots 45-52 réalisés dans ces indications aient mesuré l’impact du trastuzumab sur la survie globale sur des durées beaucoup plus prolongées que dans le cancer métastatique, les gains absolus de survie sont encore plus faibles, voire inexistants dans quelques-unes de ces études. Il en résulte une proportion encore plus importante de femmes traitées sans allongement de leur vie.
Un essai clinique randomisé ouvert conduit chez 3222 patientes atteintes dans cancer du sein HER+ réséqué a comparé trois protocoles de chimiothérapie adjuvante dont deux incluant le trastuzumab en association 45. Les taux de survie à 5 ans sont respectivement de 87% (chimiothérapie seule) ; 92% et 91% (chimiothérapie + trastuzumab). Les gains absolus de survie à 5 ans de suivi sont donc de 5% et 4%. Les nombres de patientes à traiter pour éviter un décès supplémentaire par rapport à la chimiothérapie sont donc respectivement de 20 et 25 à 5 ans de suivi. Autrement dit, 95% et 96% des patientes sont traitées sans gain de survie.
Dans l’essai HERA 46, 47, 48 , 5081 femmes ont été randomisées dans 3 groupes, chimiothérapie adjuvante seule, ou suivie d’une injection de trastuzumab toutes les 3 semaines pendant un an, ou pendant 2 ans. Le gain absolu de survie après deux ans de suivi 46 est de 0,9% et non significatif (95,1% versus 96% de survivantes respectivement pour la chimiothérapie adjuvante seule par rapport à l’ajout de trastuzumab). En revanche, il y a 8,5% d’arrêt de traitement dans le groupe trastuzumab contre aucun de signalé dans le groupe d’observation, 7,9% versus 4,4% de patientes qui subissent au moins un événement indésirable de grade 3 ou 4 (p<0,001), 7,0% versus 4,7% de patientes qui présentent au moins un événement indésirable sévère (p=0,007), et la toxicité cardiaque du trastuzumab est avérée : 29 patientes (1,7%) versus 1 (0,06%) présentent une insuffisance cardiaque congestive symptomatique (p< 0,001), dont 9 sévères (0,54%) versus 0 (p=0,002), et 7,1% versus 2,2% des patientes qui présentent une diminution de leur fraction d’éjection ventriculaire gauche (p< 0,001).
Après un suivi médian de 8 années 47, deux années de trastuzumab ne font pas mieux qu’une seule sur la survie globale (voir la figure S3 à retrouver dans le supplément).
Sur l’ensemble de la population incluse dans l’étude, les taux de survie à 12 ans 48 sont respectivement de 72,9% (chimiothérapie adjuvante seule), 79,4% (trastuzumab pendant un an) et 79,5% (trastuzumab pendant 2 ans). Le gain absolu de survie pour le groupe un an de trastuzumab par rapport à la chimiothérapie adjuvante seule est de 6,5%. Ainsi, 93% des patientes sont traitées inutilement pendant 12 années sans gain de survie. Le bénéfice est encore plus faible pour le sous-groupe des femmes hormonosensibles avec un gain absolu de survie après 12 ans de 4,7%, soit un NNT de 21, et donc plus de 95% de patientes traitées inutilement sans gain de survie !
En revanche, le traitement par trastuzumab fait plus que doubler la proportion de patientes qui subissent au moins un événement indésirable sévère (grade 3 ou 4) qui passe de 8,7% (chimiothérapie adjuvante seule) à 17,5% (un an de trastumuzab) et à 21,8% (deux ans de trastuzumab). Il triple presque les décès liés à un événement indésirable (respectivement 0,6%, 1,6% et 1,6%) et augmente très fortement la fréquence des insuffisances cardiaques congestives (Voir le tableau S7, page 14 du supplément).
Deux essais cliniques randomisés conduits chez 4046 femmes atteintes d’un cancer du sein opérable 49, 50, 51, ont fait l’objet d’une analyse combinée. Les taux de survie calculés à dix ans sont respectivement de 75,2% et 84, soit un gain absolu de survie de 8,8%. Le NNT est donc de 11. Il en résulte donc que 91% des patientes sont traitées pendant 10 ans sans gain de survie
Enfin, dans l’essai NOAH 52, le gain de survie globale (74% versus 63%) n’est pas statistiquement significatif)
Mais, revenons aux prix de ces nouveaux anticancéreux qui s’envolent :
De nouveaux sommets ont récemment été atteints avec le KYPROLIS®, carfilzomib, AMGEN, admis au remboursement en juillet 2018, en triple combinaison avec REVLIMID®, lénalidomide, CELGENE, déjà fort coûteux, et la dexaméthasone, dans le myélome multiple chez des patients adultes ayant reçu au moins un traitement antérieur, selon les résultats de l’étude ASPIRE après 67,1 mois de suivi (5,6 années) dans un essai qui comparait donc cette triple association au REVLIMID® + dexaméthasone (J Clin Oncol 36 ;8:728-734. Online January 17, 2018 En accès libre : ici).
Le gain absolu de survie globale après 5,6 années de suivi n’est que de 5,3%, il faut traiter 19 patients (NNT) pour éviter un seul décès au surcoût de près de 5 millions d’euros par rapport au comparateur actif.
Dans le même temps, 87% des patients traités par carfilzomib subissent un effet indésirable de grade 3 ou supérieur, c’est-à-dire ayant entraîné une hospitalisation, des séquelles, ou menacé la vie du patient.
Ces modes d’expression des résultats des essais cliniques (gain absolu de survie, nombre de patients à traiter et coût supplémentaire pour éviter un décès par rapport à un comparateur actif), cruciaux pour une information loyale des patients et des assurés sociaux, ne figurent nulle part. Vous ne les trouverez ni sur l’EPAR de l’EMA (ici), ni sur l’avis du 21 février 2018 de la Commission de la transparence de la HAS (ici), ni sur le site de l’INCa, ni sur la base de données publique du médicament (ici) sensée permettre au grand public et aux professionnels de santé d’accéder à des données et documents de référence. Une base mise en œuvre par l’ANSM, la HAS et l’UNCAM, sous l’égide du Ministère de la santé.
Dans ces conditions, comment voulez-vous que les patients puissent prendre des décisions éclairées et partagées en fonction de leurs préférences et attentes selon une perception, la plus juste possible, du bénéfice en termes de gain de survie à attendre du traitement ?
Et cette absence d’information dans ce type de format utile à la décision partagée concerne en définitive tout nouvel anticancéreux, voire tout médicament récemment mis sur le marché.
Retour
Conclusion (2ème volet) :
A ce stade, plusieurs questions se posent :
Quel intérêt auraient les patients à accéder encore plus vite à ces soit-disant innovations alors qu’ils sont si nombreux déjà à être surtraités inutilement sans gain de survie ni de qualité de vie ?
Quel intérêt aurait notre système de santé à rembourser encore plus vite et plus longtemps, et dans l’ignorance totale de leurs effets positifs comme négatifs, des médicaments aux prix exorbitants ?
Quel autre secteur industriel est autorisée à commercialiser à des prix aussi astronomiques une production aussi défectueuse ?
A quelle autre industrie l’État garantit-il une hausse annuelle de son chiffre d’affaires ?
Notre Ministre de la santé, Agnès BUZYN, déclarait le 15 décembre 2017 : « On peut rassurer les français, aujourd’hui il n’y a pas un citoyen français qui n’a pas accès à un médicament parce qu’il serait trop cher. Pas un ! Et ça va continuer. C’est mon engagement. On n’est pas l’Angleterre… Tous les français accéderont à tous les médicaments s’ils sont utiles quelque soit le prix ». Utiles pour qui ?
Revoir l’interview d’Agnès BUZYN calée sur cette déclaration solennelle. Cliquer sur l’image ci-dessous pour lancer la vidéo


Volet 3 – Petite revue de presse
Le discours sur les « médicaments innovants » dans les médias comme au Sénat semble reprendre en grande partie les éléments de langage d’une source unique:
une conférence de presse du LEEM, le syndicat de l’industrie pharmaceutique française, qui présentait son « Plan cancer » et lançait ainsi une année de lobbying en vue du CSIS.
Y intervenaient, à l’appui du LEEM, l’Institut Curie et l’Institut Gustave Roussy, que l’on retrouvera ensuite dans les médias et commissions parlementaires.
- Au journal télévisé de 20h de France 2, le 20 juin 2018 : « Ce qui fait débat : Des médicaments trop chers ? »
Cliquer sur l’image ci-dessous puis sur la petite flèche pour visionner l’extrait vidéo du JT (4 min 08 s)
A 2 min 08 s de l’enregistrement, le Pr Jean-François MORÈRE, oncologue à l’AP-HP, répond à la question : « Leur prix est-il justifié ? »
« Aujourd’hui l’immunothérapie permet des réponses du cancer qui n’ont jamais été observées avec une telle rapidité avec les traitements classiques. C’est-à-dire que vous pouvez après deux injections avoir une quasi disparition totale de la maladie ».
Le Pr MORÈRE ne précise pas que ces effets décrits comme miraculeux ne concernent qu’un nombre extrêmement restreint de patients, la plupart des malades traités ne bénéficiant d’aucun gain de survie. Ce discours, problématique en ce qu’il attise des espoirs qui pour la plupart seront déçus, ne respecte pas non plus la législation.
L’article L. 4113-13 du Code de la santé publique précise en effet que « les membres des professions médicales qui ont des liens avec des entreprises et établissements produisant ou exploitant des produits de santé ou des organismes de conseil intervenant sur ces produits sont tenus de les faire connaître au public lorsqu’ils s’expriment lors d’une manifestation publique ou dans la presse écrite ou audiovisuelle sur de tels produits.». Dans le cas du Pr Morère, ces liens sont nombreux:
Cliquer sur l’image ci-dessous pour ouvrir le fichier d’analyse détaillée des conflits d’intérêts du Pr. Jean-François MORÈRE, qui s’est exprimé sur France 2
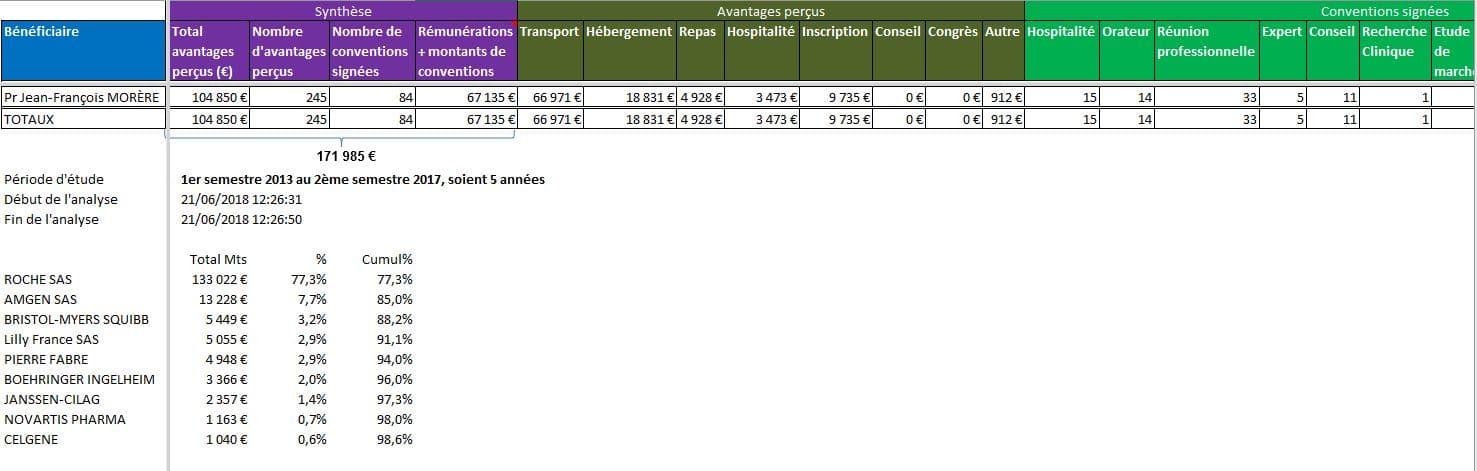
Les données de la base transparence nous apprennent que les firmes ont déclaré pour cet oncologue en avantages, conventions et rémunérations sur 5 années, la somme de 171.985 €. Parmi les entreprises rémunératrices, nous retrouvons les principaux industriels concernés par ces innovations dites de « rupture » sur lequel le professeur est interviewé.
Retour
- Le point de vue de l’économiste, Pr Claude LE PEN: « Pour l’instant ça passe »
Cliquer sur l’image ci-dessous pour lancer l’ensemble de l’enregistrement vidéo (4 min 08 s)
« Pour l’instant ça passe » nous rassure (à moitié) M. Claude LE PEN. Professeur d’économie de la santé à l’Université Paris Dauphine (ici), Claude LE PEN est surtout un consultant très actif, en premier lieu pour les industriels du médicament et des dispositifs médicaux, mais aussi pour la plupart des acteurs de la santé. En 2004, son cabinet de conseils, CLP-Santé a fusionné avec celui d’Annie CHICOYE, une autre économiste de la santé, pour former le cabinet AREMIS (ici). Au moment de le revendre, pour un montant gardé confidentiel, à IMS Health deux ans plus tard, les deux associés déclaraient 3,630 millions d’euros de chiffre d’affaires annuel. IMS-Health est le leader mondial des données de santé, devenu après sa fusion avec Quintiles IQVIA, numéro un des CRO (organismes de recherche sous contrat, auxquels l’industrie pharmaceutique sous-traite sa recherche). Claude LE PEN continue depuis le rachat à travailler avec cette entreprise (ici ).
Claude LE PEN préside également le « Cercle des Economistes de la Santé ». Il est utile là encore de préciser la nature de ce cercle, qui réunit économistes, mais également institutions et entreprises. Les 29 firmes de santé membres à part entière du Cercle des Economistes de la Santé lui assurent 60% de ses cotisations, sans compter d’éventuels concours ponctuels à l’occasion de salons, congrès ou publications. Là encore, la frontière entre expertise et lobbying est difficile à tracer.
Retour
- France Info diffusait sur ses ondes le 4 février 2018 les propos du Pr. François-Clément BIDARD, oncologue à l’Institut Curie
Cliquer sur l’image ci-dessous pour écouter le reportage diffusé par France Info (3 min 50 s)
« Cette prise de sang nous permet de chercher des mutations qui nous permettent parfois d’orienter des patients vers des traitements auxquels nous n’aurions pas pu penser sans cette prise de sang, et ça par contre, notamment dans le cancer du poumon, c’est déjà en utilisation quotidienne en France depuis maintenant un à deux ans »
Là encore la loi n’a pas été respectée, car les liens d’intérêts du jeune professeur n’ont pas été mentionnés. Ils sont pourtant déjà conséquents pour cet oncologue:
Cliquer sur l’image ci-dessus pour télécharger le fichier d’analyse. Voir aussi ses déplacements dans l’onglet du même nom…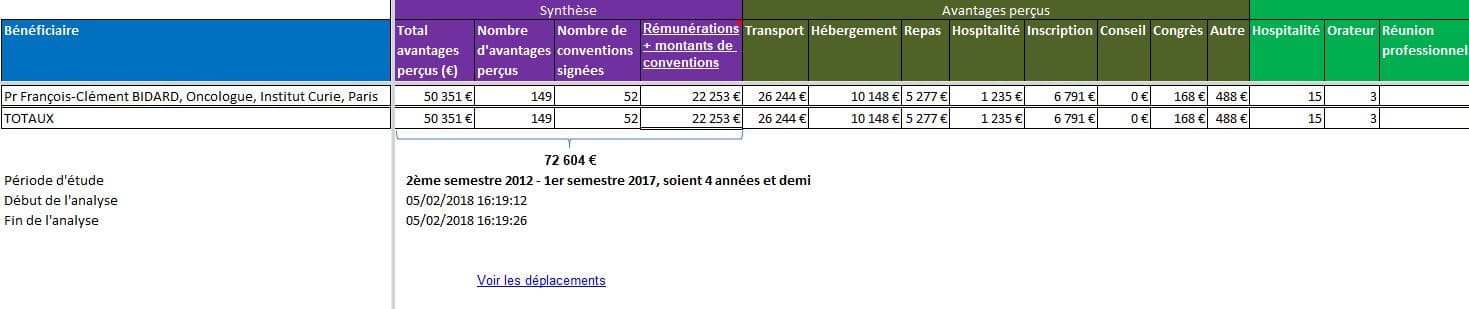
Retour
- Le marketing politique
Le Républicain Lorrain, titrait le 22 juillet 2018 « Médicaments innovants : l’urgence » (ici)
Cliquer sur l’image ci-dessous pour ouvrir l’article du Républicain Lorrain promouvant le rapport sénatorial

En sous-titre : « Une sénatrice lorraine alerte, dans un rapport, sur la difficulté pour les patients atteints d’un cancer de bénéficier de médicaments innovants. Le Premier ministre vient de s’engager à lever certains freins ».
Remarquons les termes employés dans l’article : « Big Bang en oncologie », « mouvement de forte accélération des innovations », « arrivée dans notre pays d’anticancéreux personnalisés »… Ils sont bien représentatifs de ce que l’on peut lire le plus souvent aujourd’hui dans les média.
Retour
A titre d’exemple, petite rétrospective d’articles parus dans un seul journal, Les Echos, autour du Comité Stratégique des Industries de Santé :
06/06/2018 – « La grande victoire de l’immunothérapie » (ici). Extraits : « Impossible pour les laboratoires de compter en cancérologie si on est absent de l’immunothérapie. L’américain MERCK et le suisse ROCHE occupent les premières places. L’heure est aujourd’hui aux combinaisons de médicaments afin d’augmenter le nombre de patients susceptibles d’en bénéficier ou d’améliorer l’efficacité des traitements… Le système d’évaluation et de fixation du prix des médicaments a été conçu pour réguler une innovation courante, non pour des innovations de rupture »
03/07/2018 – Jérôme MOUMINOUX, Tribune par le Directeur Accès au marché et « Corporate Affairs » de Pfizer France, « Pour que l’évaluation des nouveaux médicaments suivent le rythme de l’innovation » (ici). Extraits : Notre système d’évaluation des médicaments innovants ralentit l’accès au progrès médical. Le risque : pénaliser les gains de chances de survie de patients atteints de maladies graves »
09/07/2018 – « Médicaments : les impasses d’un système à bout de souffle » (ici). Extraits : « Le système d’évaluation des produits et de fixation des prix n’est plus adapté à l’innovation. D’où des incohérences qui pénalisent patients et laboratoires. Une véritable usine à gaz. C’est en ces termes que nombre de laboratoires ou de médecins parlent en privé du système assurant en France l’évaluation des traitements et la fixation de leurs prix. Dans l’Hexagone, l’évaluation médico-économique d’un médicament est assurée par la Commission de transparence de la Haute autorité de santé (HAS) et la détermination des prix par le Comité économique des produits de santé (CEPS). Réformer ce… »
L’article en ligne montre sous le titre une image publicitaire (interdite normalement…) :
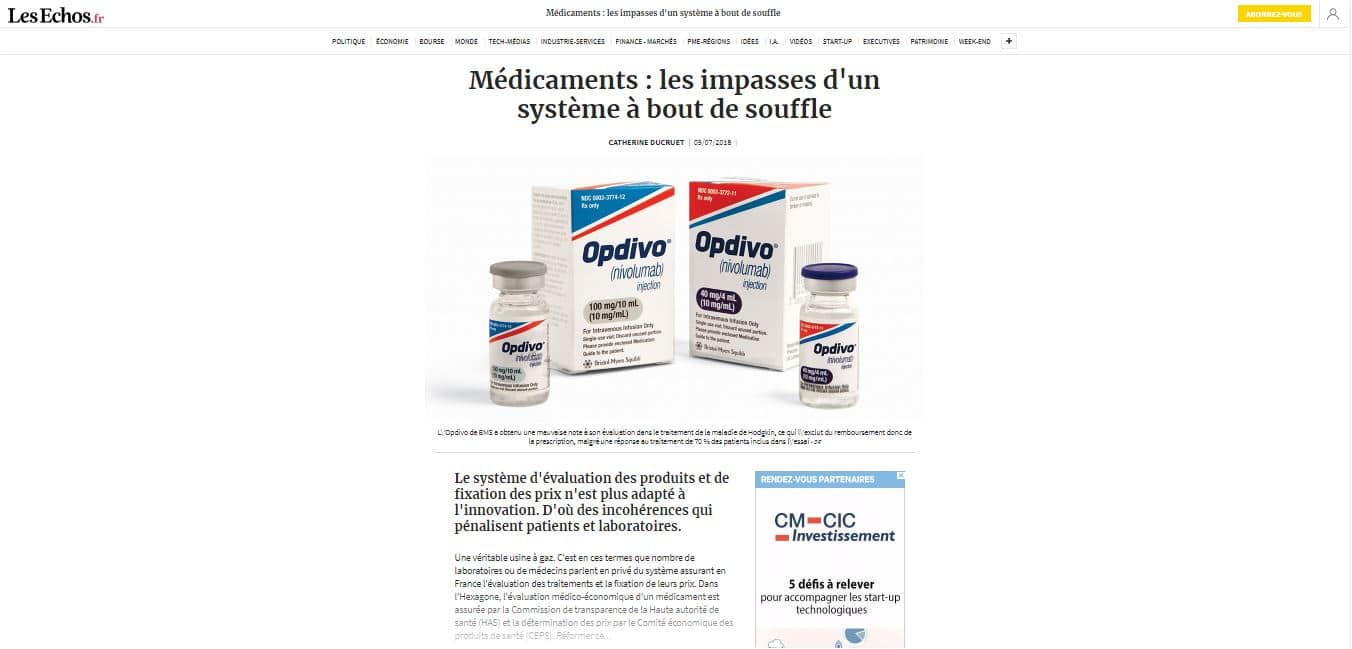
Sous l’image une légende : « L’Opdivo de BMS a obtenu une mauvaise note à son évaluation dans le traitement de la maladie de Hodgkin, ce qui l’exclut du remboursement donc de la prescription, malgré une réponse au traitement de 70 % des patients inclus dans l’essai – DR »
10/07/2018 – « Médicaments : l’exécutif veut faciliter la vie des industriels » (ici). Extraits : « Le Premier ministre a dévoilé des mesures pour l’attractivité de la France devant le conseil stratégique des industries de santé. Les délais pour autoriser les essais cliniques et fixer un prix définitif vont être comprimés. Il va falloir rattraper le temps perdu. Le Premier ministre Édouard PHILIPPE a pris la parole mardi devant le conseil stratégique des industries de santé (CSIS), pour présenter des mesures favorables aux industries du médicament et du dispositif médical »
10/07/2018 – « Olivier BRANDICOURT : « Les mesures du gouvernement sont très encourageantes » (ici). Extraits : « Dans un entretien aux « Echos », Olivier BRANDICOURT, Directeur Général de Sanofi-Aventis analyse les décisions de l’exécutif et annonce le doublement de la participation de Sanofi dans deux fonds destinés à l’innovation. Quelle est votre perception des annonces du Premier Ministre à l’issue du Conseil stratégique des industries de santé ? Les mesures annoncées ce mardi par le Premier Ministre, notamment sur le raccourcissement des délais administratifs et l’accès facilité à l’innovation pour les patients, sont très encourageantes et vont dans le bon sens. Nous avons la sensation cette fois d’une vraie volonté politique. Mes homologues des grands… »
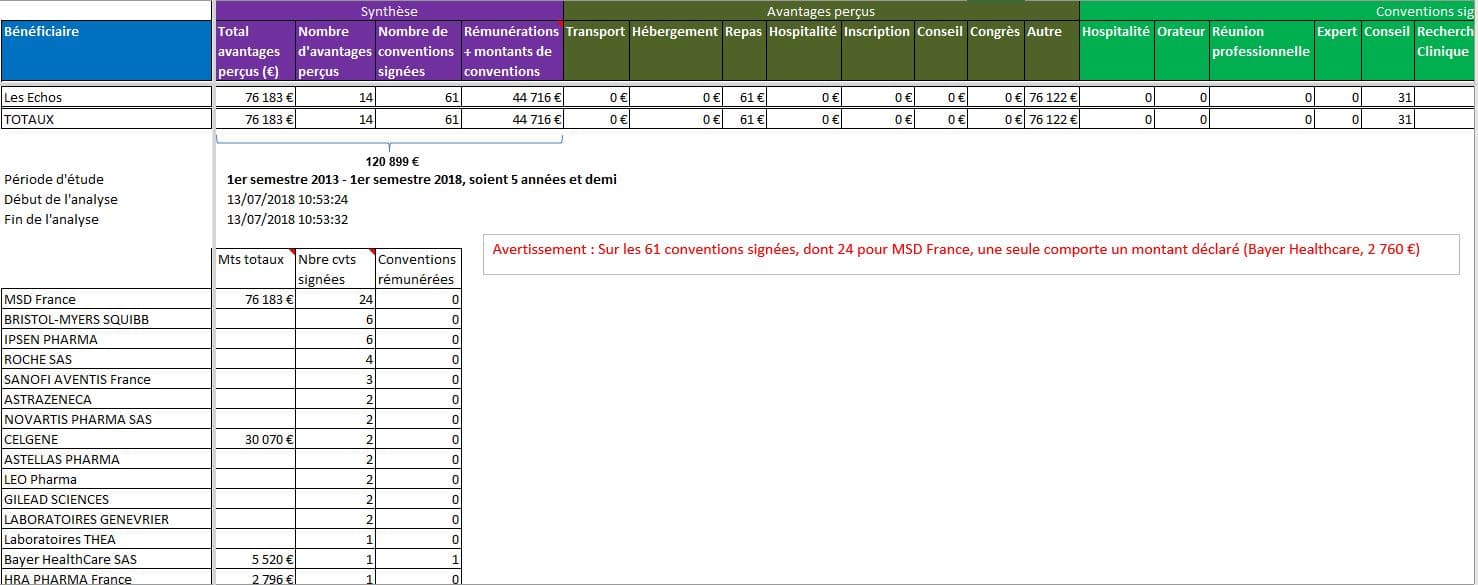
L’enthousiasme des Echos pour les « médicaments innovants » est-il libre, ou influencé par la peur de perdre le marché des laboratoires concernés? Sur la période disponible dans la base Transparence Santé, les laboratoires ont déclaré 61 contrats avec le groupe Les Echos, dont seulement 1 mentionne un montant. Les rares montants déclarés représentent plus de 120 000 EUR sur la période.
Cliquer sur l’image ci-dessous pour ouvrir le fichier d’analyse détaillée
Retour
- Une émission coproduite par « Fréquence Médicale » et « Pourquoi Docteur ? »
Dans l’émission « La santé en questions » publiée sur Youtube le 23 septembre 2018 et vue près de 135.000 fois par les internautes, l’animateur, « médecin journaliste », le Dr Jean-François LEMOINE, interviewant le Directeur général de l’assurance maladie, Nicolas REVEL, pensait surprendre son interlocuteur en le plaçant face à une situation « douloureuse » et à deux invités successifs.
Il commence par rediffuser la séquence d’une émission précédente au cours de laquelle un oncologue présent sur le plateau, lui avait permis de mettre en scène une de ses patientes, atteinte d’un cancer de la vessie métastasé et qui avait eu beaucoup de chance d’avoir bien répondu à un traitement par immunothérapie. Et l’oncologue d’affirmer qu’il ne lui serait plus possible de donner ce médicament à de nouveaux patients. Et la patiente d’implorer la ministre en invoquant des « vies à sauver ». Puis, joint par téléphone, le cancérologue répond aux questions du journaliste et maintien ses propos, en affirmant qu’il s’agit d’un scandale et que « tous les autres pays européens peuvent y avoir accès ».
Cliquer sur l’image ci-dessous pour lancer la vidéo à 26 min 39 s de l’enregistrement
Heureusement, le Directeur de la CNAMTS ne tombe pas dans le piège tendu. Le médicament en question apporte un progrès mineur selon la Haute Autorité de Santé (ASMR IV) ce qui n’a pas permis de l’inscrire sur la liste en sus. L’hôpital, alors qu’il pourrait très bien le délivrer, a choisi délibérément de ne pas le faire afin d’éviter d’avoir à financer ce traitement sur les recettes de la tarification à l’activité. Calmement mais avec fermeté, le patron de l’assurance maladie remet les pendules à l’heure : « Ce médicament est aujourd’hui disponible en France, on ne peut pas dire qu’il a été interdit. Il est même disponible dans les hôpitaux… Mais, pardonnez-moi de le dire, aujourd’hui il n’est pas vrai de dire que ce médicament est inaccessible en France, il n’est pas vrai de dire qu’un hôpital ne peut pas le délivrer ».
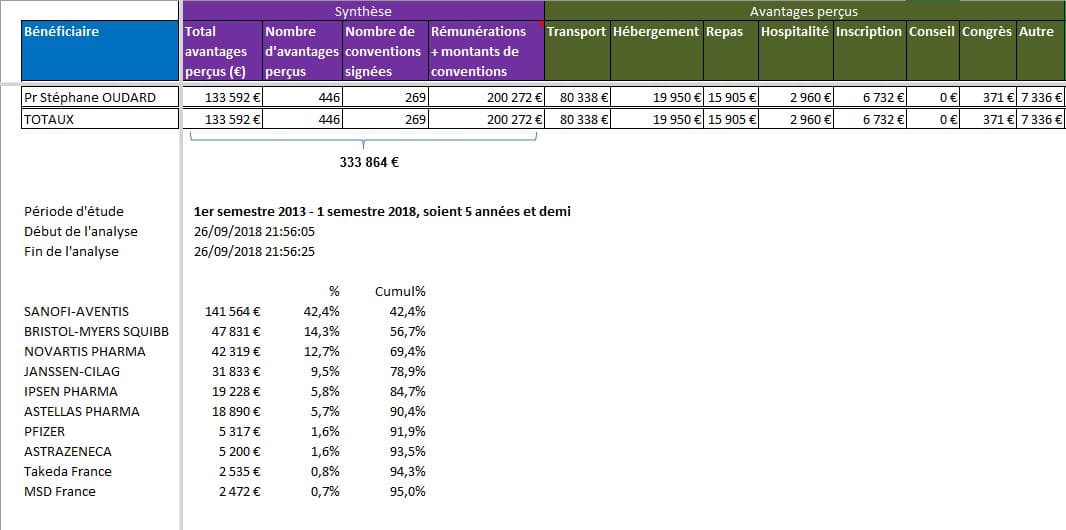
Encore une fois, les liens d’intérêts de l’oncologue, le Pr Stéphane OUDARD, n’ont pas été présentés lors de l’émission. Ils sont pourtant intéressants:
Cliquer sur l’image ci-dessous pour ouvrir le fichier d’analyse détaillée des liens d’intérêts déclarés dans la base Transparence santé
Le journaliste Jean-François LEMOINE se tourne alors vers son deuxième invité, le Dr Philippe TCHENG, nouveau Président du LEEM, et PDG du groupe Sanofi-Aventis, pour le prendre à partie et le faire réagir sur le même sujet.
Selon les éléments de langage suggérés par les communicants, celui-ci commence par ressortir du vestiaire le costume de médecin cardiologue qu’il y a rangé voici plus de 30 ans, avant d’entamer sa carrière de dirigeant dans l’industrie pharmaceutique. « D’un point de vue médical, c’est extrêmement choquant » s’exclame-t-il. Puis il tire à boulet rouge sur l’évaluation des nouveaux médicaments par la HAS, parlant de critères qu’il considère « obsolètes », et déplore les délais en France qu’il attribue à la « lourdeur et la complexité administrative ». Ces points sont vivement contestés par le Directeur de la Caisse Nationale, qui ne partage pas cet avis. La CNAMTS ayant 6 représentants au Comité Economique des Produits de Santé (CEPS), son directeur peut témoigner en toute connaissance de cause : « la négociation des prix, ça c’est quand même une responsabilité partagée, parce que lorsque l’on tarde à se mettre d’accord sur un prix, c’est parce que l’on est sur des écarts considérables, et ça c’est quand même un des sujets de la procédure d’ATU qui permet à un labo qui bénéficie de ce régime de fixer (librement) son prix pendant qu’il est en ATU. Et il sait qu’au moment où il va négocier son prix définitif, celui-ci va s’appliquer à toute la période préalable, et donc, il n’a pas tellement intérêt à ce que les choses se débouclent. Pour aller vite, il faut aussi être deux ».
Cliquer sur l’image ci-dessous pour ouvrir la vidéo à 31 min 58 s de l’enregistrement lorsqu’intervient le président du LEEM
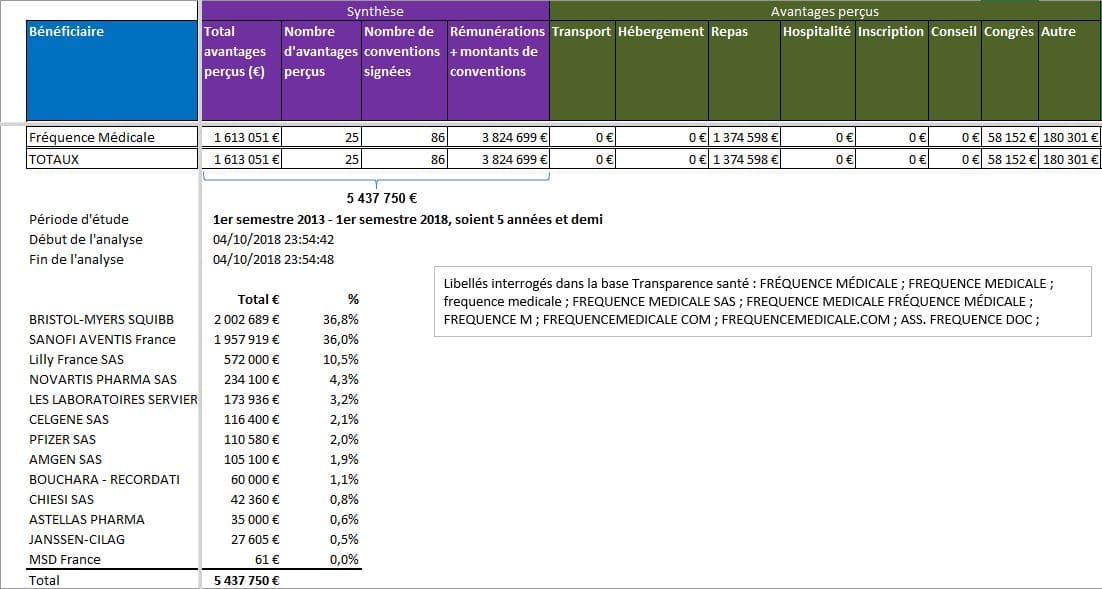
Fréquence Médicale repose sur ses prestations aux laboratoires pharmaceutiques, selon la base Transparence santé, avec plus de 5 millions d’euros déclarés versés depuis 2013:
Cliquer sur l’image ci-dessous pour télécharger le fichier d’analyse des liens déclarés pour Fréquence Médicale
Retour
- Un colloque sur les biosimilaires organisé le 28 juin 2018 par Le Quotidien du Médecin, Le Quotidien du Pharmacien, Décision & Stratégie Santé » avec le soutien de Biogaran et Sandoz
S’il existe un empire de presse qui règne en maître sur l’information médicale des médecins, tant en ville qu’à l’hôpital, c’est bien le « Groupe Profession Santé » présidé par Gérard KOUCHNER.
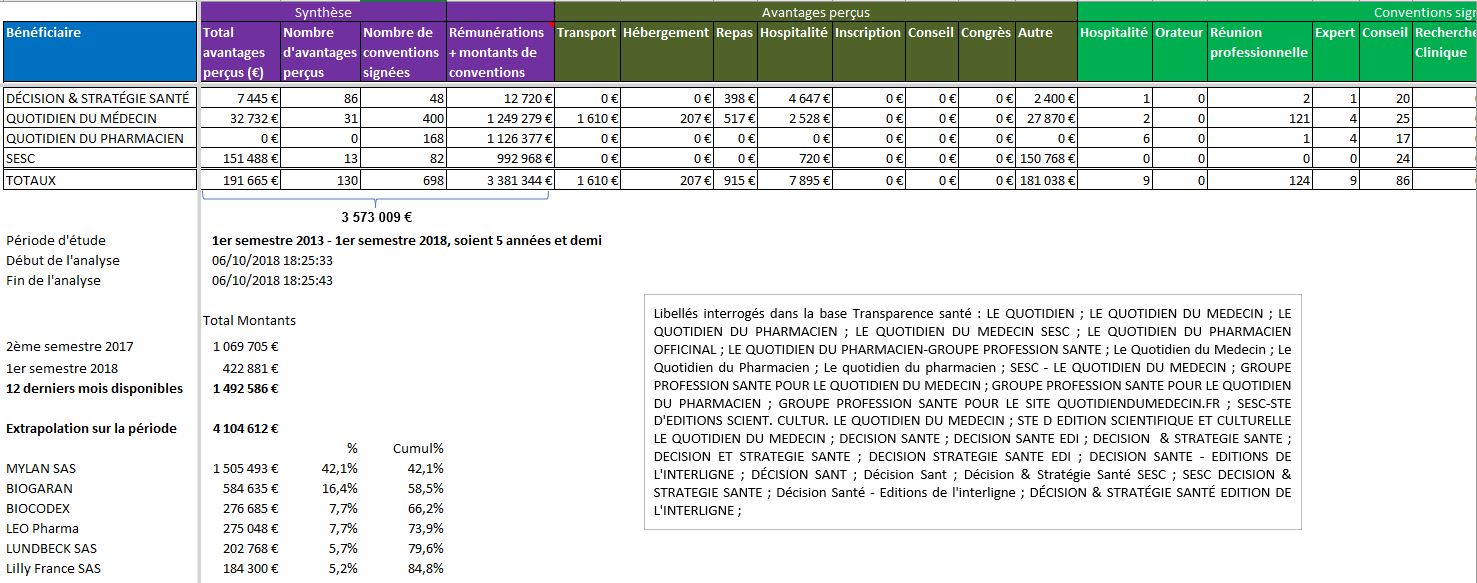
Le modèle économique des journaux médicaux du groupe de presse de Gérard KOUCHNER mérite d’être rappelé. Souvent gratuits pour les lecteurs professionnels de santé, ils sont financés principalement par la vente de prestations publicitaires (encarts, organisation d’événements). Les constats sévères du rapport de la HAS « Bonnes pratiques et critères de qualité des revues et journaux de la presse médicale française » restent hélas d’actualité. Exemple avec les déclarations faites par les firmes de santé dans la base Transparence Santé:
Cliquer sur l’image ci-dessous pour ouvrir le fichier d’analyse
Le Pr Gilles FREYER, Chef du service Oncologie Médicale au CHU Lyon-Sud, était invité lors de ce colloque à s’exprimer sur « la valeur économique des biosimilaires ». Pour schématiser, les biosimilaires sont aux biothérapies (médicaments d’origine biologique) ce que les génériques sont aux médicaments issus de la chimie de synthèse (Pour plus d’information sur leur définition, consulter les sites de la HAS et/ou de l’ANSM). Or, ce colloque se tenait quelques semaines seulement avant l’arrivée sur le marché de 3 biosimilaires de l’HERCEPTIN®, trastuzumab, du Laboratoire ROCHE : HERZUMA® (BIOGARAN), KANJINTI® (AMGEN) et ONTRUZANT (MSD) commercialisés respectivement les 30 juillet, 30 août et 18 septembre 2018.
Cliquer sur l’image et démarrer l’enregistrement vidéo à 11 min 36 s

« Je vais être assez provoc’, moi, je suis cancérologue, je parlerai en l’occurrence du trastuzumab, puisque c’est l’actualité pour la cancérologie. J’ai fait partie de ceux qui au début des années 2000 ont prescrit un médicament qui s’appelait l’HERCEPTIN®, donc le trastuzumab, qui donc permet de guérir, ça a été une révolution, des malades, 15 à 20% des cancers du sein qui ont une petite particularité, ce que l’on appelle la surexpression de HER2 ».
Puis il poursuit :
« La France a été parmi les 3 premiers pays au monde à mettre ce médicament à disposition des patientes. Aujourd’hui, nous avons régressé dans la petite moyenne des pays de l’OCDE en termes de délais, et aujourd’hui, je suis un cancérologue qui se situe dans les 400 jours entre la connaissance de l’efficacité du médicament et la possibilité du remboursement et de l’accessibilité pour les patients, là où l’Union Européenne recommande 180 jours. Et c’est cette régression de l’accès à l’innovation aujourd’hui pour les nouveaux produits. Je peux citer un nombre important de produits qui ne sont tout simplement plus accessible aujourd’hui à l’assuré social français en cancérologie. Et c’est dans ce contexte-là que nous voyons arriver les biosimilaires et au travers de la présentation que je vais vous proposer, je plaide pour une sorte de cercle vertueux, qui est un cercle de responsabilisation, d’information des patients et transparence des politiques publiques »
Cet oncologue leader d’opinion déroule mot pour mot la rhétorique des industriels du médicament, reprise par nos sénateurs, puis par le Premier Ministre lors de l’inauguration du CSIS.
Plus loin, concluant son exposé sur la valeur des biosimilaires en cancérologie, il affirme, un peu agacé « Personnellement, je ne crois pas du tout à la valeur de l’incitation économique à l’hôpital pour l’hospitalier que je suis. Je n’ai pas du tout envie d’être incité économiquement à me transformer en un décideur économique de santé. Moi je crois dans le fonctionnement en réseau(x), dans la collaboration médico-administrative. Dans mon CHU ça marche plutôt pas mal. Et que l’on permette à ma spécialité de se développer de manière harmonieuse sans parler d’économie, ni me transformer moi en un régulateur. Je pense que je ne suis pas là pour ça ».
Sa conclusion (à 49 min 07 s de l’enregistrement), pose question :
« Moi je voudrais conclure en plaidant pour un « new deal ». Et un peu en vous disant ce que pensent la plupart des cancérologues aujourd’hui. Dans les années qui viennent, il va probablement falloir investir 1 milliard 2 par an si l’on veut garantir l’accès à l’innovation en cancérologie. Ça peut paraitre beaucoup, mais on met 4 milliards d’euros dans les bons de transport. Et la prise en charge du cancer c’est 10 à 11% des dépenses de l’assurance maladie. Donc, le cancer ne coûte pas cher… L’élément central, nous avons tous la sensation que notre système d’évaluation de la valeur des médicaments en cancérologie est à bout de souffle. Et qu’il faut le réformer. Madame BUZYN a demandé une étude, enfin du moins, une révision des procédures d’évaluation, qui montre effectivement qu’elle a conscience des difficultés du système aujourd’hui. Et il y a plein de raisons pour lesquelles il faut changer ce système probablement, du moins le repenser intégralement et si j’ose dire, l’histoire des biosimilaires tombe très bien. Par ce que nous avons là un sujet de réflexion commune, qui peut être un des éléments partie prenante de ce new deal que nous appelons de nos vœux. Et si cette réflexion globale sur la valeur des médicaments en cancérologie était actée, je pense que beaucoup de cancérologues seraient enthousiastes, en tous cas pour y participer »
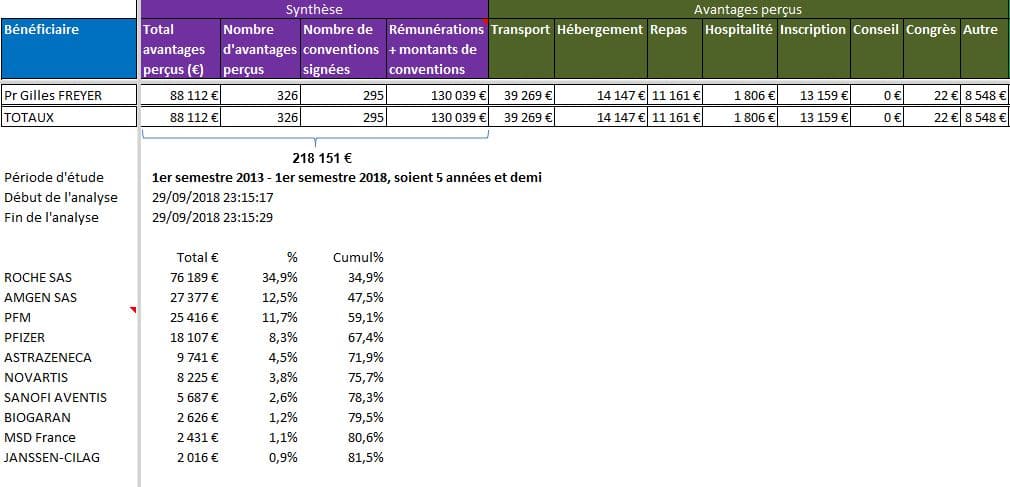
Là encore, il aurait été utile de savoir d’où parle cet oncologue. Le Pr Freyer présente de très nombreux liens d’intérêts selon la base transparence santé: pas moins de 695 déclarations enregistrées, pour un total de plus de 218 000 euros d’avantages déclarés, de montants de conventions et de rémunérations, provenant en premier lieu du laboratoire Roche dont le produit est actuellement visé par les biosimilaires.
Cliquer sur l’image ci-dessous pour ouvrir l’analyse détaillée des liens d’intérêts du Pr Gilles FREYER
On retrouve le Pr Gilles FREYER trois mois plus tard parmi les 16 signataires d’une tribune diffusée par le quotidien Le Monde, intitulée « Nos voisins européens offrent aux malades du cancer un meilleur accès à l’innovation » (ici). Il était déjà présent comme six autres de ses co-signataires dans un collectif de 78 oncologues constitué en avril 2014 pour prendre la défense d’AVASTIN®, bévacizumab, une autre thérapie ciblée des laboratoires ROCHE, contre un journaliste du magazine Le Point. L’occasion de relire l’article « Les leaders d’opinion d’une firme pharmaceutique influencent la rédaction du Point » (ici)
Retour
- Pourquoi l’innovation « de rupture » coûte cher, très cher : des éléments de réponse
Le prix des produits n’est plus fixé en relation avec les coûts de développement et de fabrication, auxquels serait ajoutée une marge, même confortable. Comme une enquête parlementaire américaine l’a montré dans le cas du Sovaldi, le prix peut aujourd’hui être fixé en sondant les patients, les médecins, les médias, les assureurs, le gouvernement sur le prix maximal au delà duquel le laboratoire risque de provoquer une révolte.
Cliquer sur l’image ci-dessous pour ouvrir la vidéo de l’enquête diffusée dans Envoyé spécial

Retour
Retour
Bibliographie :
[1] Austin B. FRAKT. The Risks and Benefits of Expedited Drug Reviews. JAMA. 2018; 320(3):225-226. doi: 10.1001/jama.2018.8262. Accès libre : ici.
[2] Scott R. BAUER, Rita F. REDBERG. Improving the Accelerated Pathway to Cancer Drug Approvals. Editor’s Note. JAMA Intern Med. 2017;177(2):278. doi:10.1001/jamainternmed.2016.7777 Accès libre : ici.
[3] Rita F. REDBERG. Faster Drug Approvals Are Not Always Better and Can Be Worse. . Editor’s Note. JAMA Intern Med. 2015;175(8):1398. doi:10.1001/jamainternmed.2015.2857 Accès libre : ici.
[4] R. ALTA CHARO. The Complexity of Integrating Speed and Safety in Drug Development and Approval. JAMA Intern Med. 2013;173(13):1165-1166. doi:10.1001/jamainternmed.2013.7161 Accès : ici.
[5] Walid F. GELLAD, Aaron S. KESSELHEIM. Accelerated Approval and Expensive Drugs – A Challenging Combination. N Engl J Med 2017; 376:2001-2004 DOI: 10.1056/NEJMp1700446 Accès libre : ici.
[6] Sana R MOSTAGHIM et al. Safety related label changes for new drugs after approval in the US through expedited regulatory pathways: retrospective cohort study. BMJ 2017;358:j3837 Accès libre : ici.
[7] Donald W LIGHT, Joel LEXCHIN. Why do cancer drugs get such an easy ride? Rushed approvals result in a poor deal for both patients and cancer research. BMJ 2015;350:h2068 doi: 10.1136/bmj.h2068 Accès libre : ici.
[8] Fiona GODLEE. Why drug approval needs better evidence. Editor’s Choice. BMJ 2016;353:i3483 doi: 10.1136/bmj.i3483 Accès libre : ici.
[9] Robert M. CALIFF. Benefit-Risk Assessments at the US Food and Drug Administration – Finding the Balance. JAMA. 2017;317(7):693-694. doi:10.1001/jama.2017.0410 Accès : ici.
[10] Alberto SOBRERO, Paolo BRUZZI. Incremental Advance or Seismic Shift? The Need to Raise the Bar of Efficacy for Drug Approval. J Clin Oncol 27;35:5868-73. Accès libre : ici.
[11] Igho J ONAKPOYA et al. Delays in the post-marketing withdrawal of drugs to which deaths have been attributed: a systematic investigation and analysis. BMC Medicine (2015) 13:26 DOI 10.1186/s12916-014-0262-7 Accès libre : ici.
[12] Tracy RUPP, Diana ZUCKERMAN. Quality of Life, Overall Survival, and Costs of Cancer Drugs Approved Based on Surrogate Endpoints. JAMA Intern Med. 2017;177(2):276-277. doi:10.1001/jamainternmed.2016.7761 Accès libre : ici.
[13] Chul KIM, Vinay PRASAD. Cancer drugs approved on the basis of a surrogate end point and subsequent overall survival: an analysis of 5 years of US Food and Drug Administration approvals. JAMA Intern Med. 2015;175(12):1992-1994. JAMA Intern Med. 2015;175(12):1992-1994. doi:10.1001/jamainternmed.2015.5868 Accès libre : ici.
[14] Rita BANZI et al. Approvals of drugs with uncertain benefit–risk profiles in Europe. European Journal of Internal Medicine 26 (2015) 572–584 Accès libre : ici.
[15] Sham MAILANKODY, Vinay PRASAD. Five Years of Cancer Drug Approvals: Innovation, Efficacy, and Costs. JAMA Oncol. 2015;1(4):539-540. doi:10.1001/jamaoncol.2015.0373 Accès libre : ici.
[16] Alain BRAILLON. Surrogate end points for overall survival. Festina lente (more haste, less speed). Annals of Oncology 26: 818–821, 2015 Accès libre : ici.
[17] Courtney DAVIS et al. Availability of evidence of benefits on overall survival and quality of life of cancer drugs approved by European Medicines Agency: retrospective cohort study of drug approvals 2009-13. BMJ 2017;359:j4530 doi: 10.1136/bmj.j4530 Accès libre : ici.
[18] Vinay PRASAD. Do cancer drugs improve survival or quality of life? You don’t need to know, according to our broken regulatory system. Editorials. BMJ 2017;359:j4528 doi: 10.1136/bmj.j4528 Accès libre : ici.
[19] Deborah COHEN. Cancer drugs: high price, uncertain value. A study published in The BMJ this week shows how most new cancer drugs are failing to deliver any clinically meaningful benefit. It’s time for Europe to raise the evidence bar before market approval, finds Deborah Cohen. BMJ 2017;359:j4543 doi: 10.1136/bmj.j4543 Accès libre : ici.
[20] Emma ROBERTSON. Patient commentary: the current model has failed. BMJ 2017;359:j4568 doi: 10.1136/bmj.j4568 Accès libre au résumé : ici.
[21] Vinay PRASAD et al. The Strength of Association Between Surrogate End Points and Survival in Oncology – A Systematic Review of Trial-Level Meta-analyses. JAMA Intern Med. 2015;175(8):1389-1398. doi:10.1001/jamainternmed.2015.2829 Accès libre : ici.
[22] Alain BRAILLON, David B.MENKES. Balancing Accelerated Approval for Drugs With Accelerated Withdrawal. JAMA Intern Med. 2016;176(4):566-567. doi:10.1001/jamainternmed.2016.0351 Accès : ici.
[23] US GAO (United States Government Accountability Office). New Drug Approval – FDA Needs to Enhance Its Oversight of Drugs Approved on the Basis of Surrogate Endpoints – Report to the Ranking Member, Committee on Finance, U.S. Senate. September 2009. Accès libre : ici.
[24] US GAO (United States Government Accountability Office). Drug Safety – FDA Expedites Many Applications, But Data for Postapproval Oversight Need Improvement – Report to the Ranking Member, Subcommittee on Labor, Health and Human Services, Education, and Related Agencies, Committee on Appropriations, House of Representatives. December 2015. Accès libre : ici.
[25] Tito FOJO et al. Unintended Consequences of Expensive Cancer Therapeutics – The Pursuit of Marginal Indications and a Me-Too Mentality That Stifles Innovation and Creativity – The John Conley Lecture. JAMA Otolaryngol Head Neck Surg. 2014;140(12):1225-1236. doi:10.1001/jamaoto.2014.1570 Accès au résumé : ici.
[26] Huseyin NACI et al. Timing and Characteristics of Cumulative Evidence Available on Novel Therapeutic Agents Receiving Food and Drug Administration Accelerated Approval. The Milbank Quarterly, Vol. 95, No. 2, 2017 (pp. 261-290). Accès libre : ici.
[27] Matthew V. ABOLA, Vinay PRASAD. The Use of Superlatives in Cancer Research. JAMA Oncol. 2016;2(1):139-141. doi:10.1001/jamaoncol.2015.3931 Accès libre : ici.
[28] Aaron P. MITCHELL et al. Pharmaceutical Industry Payments and Oncologists’ Selection of Targeted Cancer Therapies in Medicare Beneficiaries. JAMA Intern Med. 2018;178(6):854-856. doi:10.1001/jamainternmed.2018.0776 Accès : ici.
[29] Vinay PRASAD et al. The high price of anticancer drugs: origins, implications, barriers, solutions. Nature Reviews Clinical Oncology volume 14, pages 381–390 (2017) Accès au résumé : ici.
[30] Finlay Mc ALISTER. The “number needed to treat” turns 20 – and continues to be used and misused. CMAJ 2008; 179(6):549-53. Accès libre : ici.
[31] Pierre CHEVALIER. FMC : Concept et outils en EBM – Nombre de sujets à traiter. Minerva 2009 8(2) :24. Accès libre : ici.
[32] Akl EA et al. Using alternative statistical formats for presenting risks and risk reductions. Cochrane Database Syst Rev. 2011 Mar 16;(3):CD006776. doi: 10.1002/14651858.CD006776.pub2. Accès abstract : ici.
[33] Cole WAYANT et al. Financial Conflicts of Interest Among Oncologist Authors of Reports of Clinical Drug Trials – RESEARCH LETTER – JAMA Oncology Published online August 30, 2018. Accès à l’abstract et la première page : ici.
[34] Roberto FERRARA et al. Hyperprogressive Disease in Patients With Advanced Non–Small Cell Lung Cancer Treated With PD-1/PD-L1 Inhibitors or With Single-Agent Chemotherapy. JAMA Oncol. doi:10.1001/jamaoncol.2018.3676 – Published online September 6, 2018. Accès au résumé : ici.
[35] Interview du Pr Benjamin BESSE, IGR Villejuif, l’un des auteurs de l’étude précédente [34], par le JAMA Oncology. Accès libre : ici.
[36] Adam GAFFNEY, Joel LEXCHIN. Healing an ailing pharmaceutical system: prescription for reform for United States and Canada. BMJ 2018;361:k1039 doi: 10.1136/bmj.k1039 (Published 17 May 2018) Accès : ici.
[37] D. J. SLAMON et al. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N Engl J Med, Vol. 344, No. 11 · March 15, 2001. Accès libre : ici.
[38] Michel MARTY et al. Randomized Phase II Trial of the Efficacy and Safety of Trastuzumab Combined With Docetaxel in Patients With Human Epidermal Growth Factor Receptor 2–Positive Metastatic Breast Cancer Administered As First-Line Treatment: The M77001 Study Group. Journal of Clinical Oncology 23, no. 19 (July 2005) 4265-4274. Accès libre : ici.
[39] G GASPARINI et al. Randomized phase II trial of weekly paclitaxel alone versus trastuzumab plus weekly paclitaxel as first-line therapy of patients with HER-2 positive advanced breast cancer. Breast Cancer Res Treat 2007; 101: 355-65. Accès libre : ici.
[40] B KAUFMAN et al. Trastuzumab plus anastrozole versus anastrozole alone for the treatment of postmenopausal women with human epidermal growth factor receptor 2-positive, hormone receptor-positive metastatic breast cancer: results from the randomized phase III TAnDEM study. J Clin Oncol 2009; 27: 5529-37. Accès libre : ici.
[41] J HUOBER et al. Higher efficacy of letrozole in combination with trastuzumab compared to letrozole monotherapy as firstline treatment in patients with HER2-positive, hormone-receptor-positive metastatic breast cancer – results of the eLEcTRA trial. Breast 2012; 21:27-33. Accès au résumé : ici.
[42] K. L. BLACKWELL et al. Overall Survival Benefit With Lapatinib in Combination With Trastuzumab for Patients With Human Epidermal Growth Factor Receptor 2–Positive Metastatic Breast Cancer: Final Results From the EGF104900 Study. J Clin Oncol 2012; 30:2585-2592. Accès libre : ici.
[43] S BALDUZZI et al. Trastuzumab-containing regimens for metastatic breast cancer. Cochrane Database Syst Rev 2014; 6: CD006242. Accès : ici.
[44] R K. MURTHY et al. Effect of Adjuvant/Neoadjuvant Trastuzumab on Clinical Outcomes in Patients With HER2-Positive Metastatic Breast Cancer. Cancer 2014;120:1932–8. Accès libre : ici.
[45] D. SLAMON et al. Adjuvant Trastuzumab in HER2-Positive Breast Cancer. N Engl J Med 2011;365:1273-83 (BCIRG trial). Accès libre : ici ; Supplément : ici ; Protocole : ici ; Déclaration des liens d’intérêts : ici.
[46] M. J. PICCART-GEBHART et al. Trastuzumab after Adjuvant Chemotherapy in HER2-Positive Breast Cancer (HERA trial). N Engl J Med 2005;353:1659-72. Accès libre : ici ; Suppléments : ici.
[47] A. GOLDHIRSCH et al. 2 years versus 1 year of adjuvant trastuzumab for HER2-positive breast cancer (HERA): an open-label, randomised controlled trial. Lancet 2013; 382: 1021–28. Accès libre grace à Google Scholar (ici).
[48] D. CAMERON et al. 11 years’ follow-up of trastuzumab after adjuvant chemotherapy in HER2-positive early breast cancer: final analysis of the HERceptin Adjuvant (HERA) trial. Lancet. 2017 March 25; 389(10075): 1195–1205. doi:10.1016/S0140-6736(16)32616-2. Accès libre : ici ; Supplément accessible : ici.
[49] E. A. PEREZ et al. Trastuzumab plus adjuvant chemotherapy for human epidermal growth factor receptor 2-positive breast cancer: planned joint analysis of overall survival from NSABP B-31 and NCCTG N9831. J Clin Oncol 2014;32:3744-52. Accès libre ici.
[50] E. H. ROMOND et al. Trastuzumab plus Adjuvant Chemotherapy for Operable HER2-Positive Breast Cancer. N Engl J Med 2005;353:1673-84. Accès libre : ici ; Supplément : ici.
[51] E. A. PEREZ et al. Four-Year Follow-Up of Trastuzumab Plus Adjuvant Chemotherapy for Operable Human Epidermal Growth Factor Receptor 2–Positive Breast Cancer: Joint Analysis of Data From NCCTG N9831 and NSABP B-31. J Clin Oncol 2011;29:3366-3373. Accès libre : ici.
[52] Luca GIANNI et al. Neoadjuvant and adjuvant trastuzumab in patients with HER2-positive locally advanced breast cancer (NOAH): follow-up of a randomised controlled superiority trial with a parallel HER2-negative cohort. Lancet Oncol 2014;15: 640-47. Accès libre : ici ;










Laisser un commentaire